by Olivia Smith
Welcome to “Wrangling Sequencing Data” - yeehaw! There’s a lot of different ways to tackle this, so this is an attempt at the guide to metagenomics data management that I needed when I first started. There’s a substantial amount of ground to cover so this will come in three parts. In Part 1, I’ll go into what kinds of files you might encounter (will be familiar to more experienced folks). In Part 2, I’ll discuss how to get them (hopefully helpful to most). Finally, in Part 3, I’ll briefly discuss a couple of tools for working with all these files, but I’ll leave most of the deeper investigation on those tools up to you.
How I recommend using this guide: read relevant or interesting-to-you sections as you go. There’s a lot of info here from file types to bash loops to tool recommendations, and I don’t expect all of it to be necessary or helpful for everyone.
Also. There’s a tl;dr below each section header.
Part 1: File Formats
There are 3 different kinds of files ~metagenomicists~ (you!) commonly come across. These are all text files. It’s important to know the difference between them so that you can work with them appropriately.
tl;dr Sequencing Reads: List of named reads with base call quality scores Alignment files: Map of the ordered reads in the overall sequence Index files: Complementary file which makes it less computationally intensive to scan through a big file Consensus sequence: FASTA file with list of named, ‘pretty’ sequences
Sequencing Reads
Sequencing reads (FASTQ) are the most ‘raw’ version of sequencing data that you will get. This is what the sequencing center will return after a sequencing run. These files usually have extension .fastq or .fastq.gz (.gz indicates that the file was compressed with the software tool gzip). I like to think of them as just a big alphabet soup of reads.
Reads can either be single-end (reading in one direction) or paired-end (reads going both backwards and forwards). Paired-end sequencing is more expensive, but it improves the quality of assemblies by allowing you to better place the reads in the right location. When they are paired-end, you will most often see two .fastq files (forward and backward). If so, you will want both of these files.
The file will be full of reads shown in this seemingly unintelligible format:
@ERR3654168.1 M_PC0253:13:C3N9HACXX:8:1114:7573:78875/1
TCTGCACAGATTTCGGTGGTACTCTGAAGGCGGAGCAC
+
JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ
This table shows a line-by-line breakdown of each entry (read) in the file:

†Some databases or organizations have specific formats for the headers. Wikipedia has a good explanation of this.
Line 4 is the kind of weird one (but a really important one!). The scores aren’t just numbers like you’d expect, but instead are ASCII characters. We also see this in alignment files, as noted below. The quality scores follow this pattern (increasing quality from left to right, so ! is lowest quality, ~ is highest):
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
Quality scores are very important, because it makes it possible to filter out issues in the sequencing data before you work with the files. Even with ancient sequencing data, you will want to do some form of quality control. Often when working with aDNA, the ends of the reads are trimmed (removed, due to damage often occurring there), a quality score threshold is used, and reads are constrained by length (again, since aDNA is damaged and so doesn’t generally produce the normal 250+ bp reads we see in non-degraded DNA.
Alignment Files
Once you have your sequence read files, the next step is often to create an alignment. Alignment software figures out which reads overlap and what order they should go in to recreate the DNA sequence. I’m not experienced with aligning so I won’t discuss how it works any further – see Chowdhury and Garai (2017) for a review on technical details and various tools. Alignment generally creates either a SAM or BAM file with extension .sam or .bam, respectively. Most often you will use BAM files since these are binary (compressed) versions.
If you were to open a BAM file and look at it in the terminal you would see this (command to do so is the first line):
$ samtools view OBP001.TF.bam | less
M_K00233:75:HMVJGBBXX:7:2110:5477:22590 0 1 882544 37 73M * 0 0 NNNNNNNNNNATTTGGCCCCTCACCAAAAACATTTTCTGACCCCTACCCCAGACCCCGACCCTNNNNNNNNNN !!!!!!!!!!JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ!!!!!!!!!! XT:A:U NM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:73 RG:Z:ILLUMINA-OBP001.A0101.TF1.1_S0_L007_R1_001.fastq
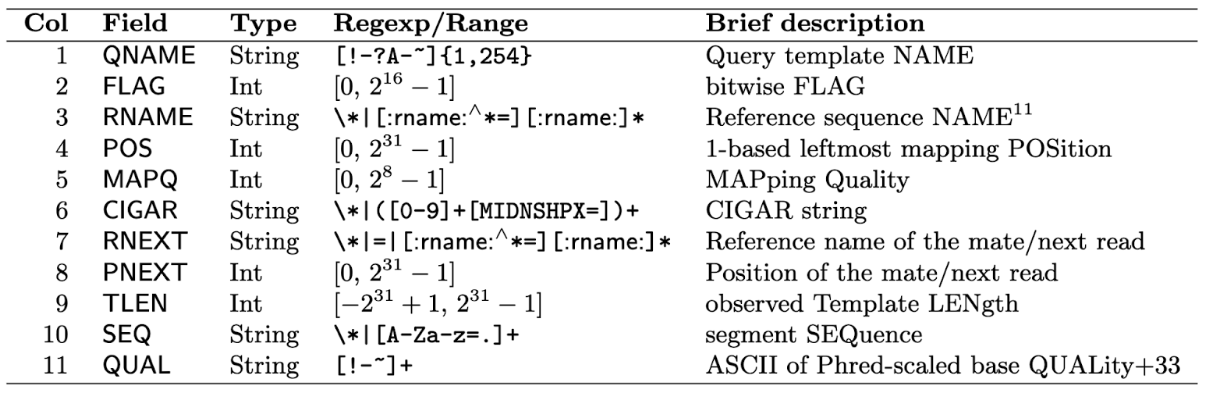
Just like with the fastq files, this isn’t really meant to be read by you! It’s meant to be read by a computer. But knowing what it should look like is helpful when you need a sanity check. It’s good practice to sanity check files when you first download them, especially if you didn’t generate them. Below is a table from the SAMTools documentation describing each column. It’s unusual that you’ll need to work with all of these, but it’s again useful to point out the quality scores for the mapping (MAPQ) and base calling (QUAL). Remember: you will use these columns for quality filtering before analysis.
Index Files
In addition to your alignment files, you’ll need a partner file called an index file. This file is important because it allows for faster searching when working with the file in SAMtools or BCFtools.
Most often when you download your BAM file, it won’t come with the index file. You can generate it very easily using:
$ samtools index OBP001.TF.bam
By default, this will create a file with the same name and .bai added to the end (in this case OBP001.TF.bam.bai). Note that it is required that the file has the correct naming convention and is in the same location as the alignment file. But just let SAMtools do its default thing and you’ll be good!
I’m not going to show you this file, because it really really doesn’t make any sense. But you can check that it’s not empty using the bash command for word count, wc. The -l flag will return the number of lines and full file path. If it returns 0 or an empty line, something is wrong.
$ wc -l OBP001.TF.bam.bai
13652 /scratch1/07869/osmith/Tutorial/OBP001.TF.bam.bai
Consensus Sequences
A consensus sequence (FASTA) is the most palatable version of a sequence and usually has the file extension .fasta. It is just the ‘refined’ sequence with headers specifying the sequence name (often a gene name, contig, or chromosome, though it can be anything).
These files look like some version of this (where each > marks a new sequence):
>NC_000020.11 Homo sapiens chromosome 20, GRCh38.p14 Primary Assembly
NNNNNNNNNNTGTTCAGTCGGGCAGGGAGTGGGAATAGACAAGACCACAAGCAGCTTGGTGCCTCTGAAAGG
GAGAGGGGTGGAGGGGAGACTAGAGAGGTGGGTAGGAATACTGGATTCCACTGACCACGTGCTGGATGTCAT
GCTTAGCCCTCCTGCTCTGTGCCAGGTTAGGCACCTGGTGTTTTACATATATTATATTACATTCTATTACAG
ACAACTCCATAGCAATCCTTTCCTCTCCATTCCATTTCTCTCCACTCCATCCCATTCCATTCCA
>NC_000021.9 Homo sapiens chromosome 21, GRCh38.p14 Primary Assembly
NNNNNNNNNNNNNNNNNNGATCCACCCGCCTTGGCCTCCTAAAGTGCTGGGATTACAG
GTGTTAGCCACCACGTCCAGCTGTTAATTTTTATTTAATAAGAATGACAGAGTGAGGGCCATCACTGTTA
ATGAAGCCAGTGTTGCTCACAGCCTCCCCTTGGTCACTTTTTGTGACTGAAGGGCATGTGTTCAGGCAAG
ATTGTTGGGTGGCTGTGTTTTGTCTTCTTCCAGCTCGGCCATGGAATAGCC
While consensus sequences are common in modern genomics (including modern metagenomics and genetic engineering), they are less often seen in paleogenomics. This is because damaged DNA makes it more challenging to confidently call the sequence.
You can also generate index files for consensus FASTA sequences using
samtools faidx {FILE}
which makes the file less computationally intensive to work with (see ‘Index Files’ above) and will generate a file with the same name plus the .faidx extension.
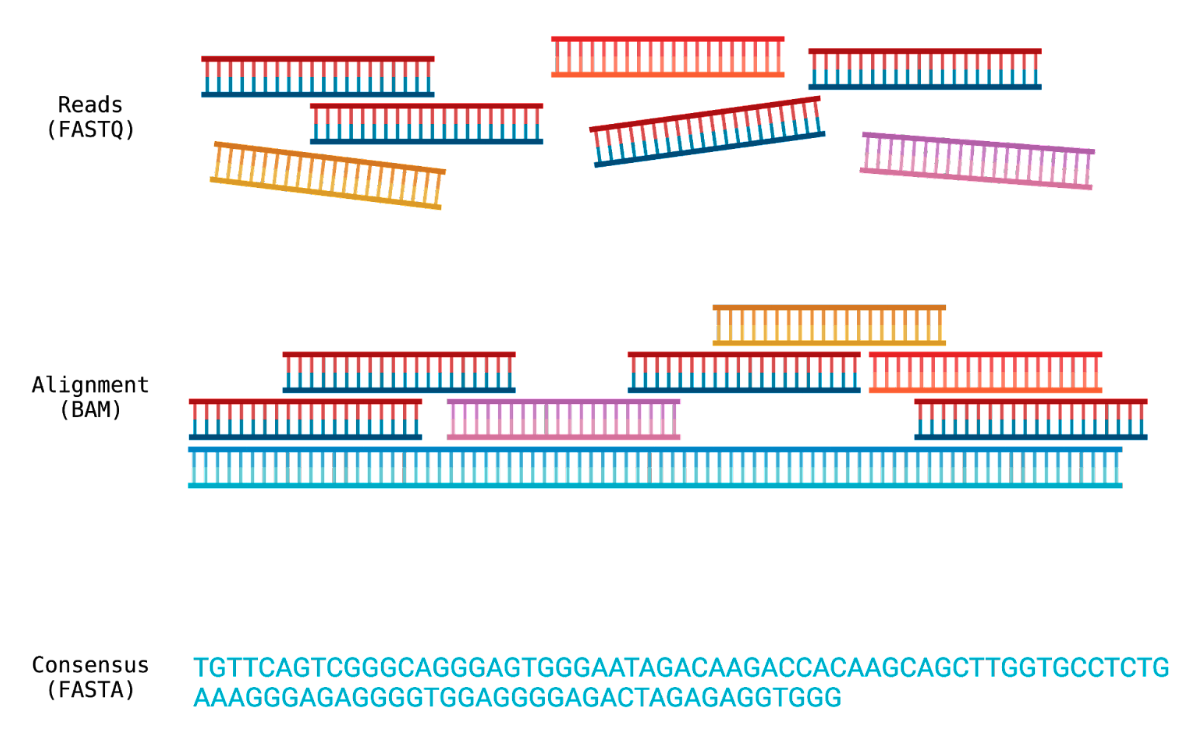
The difference between FASTQ, BAM and FASTA files. Created with Biorender.com
That’s all for Part 1, be sure to come back to read Part 2: Download Tools!