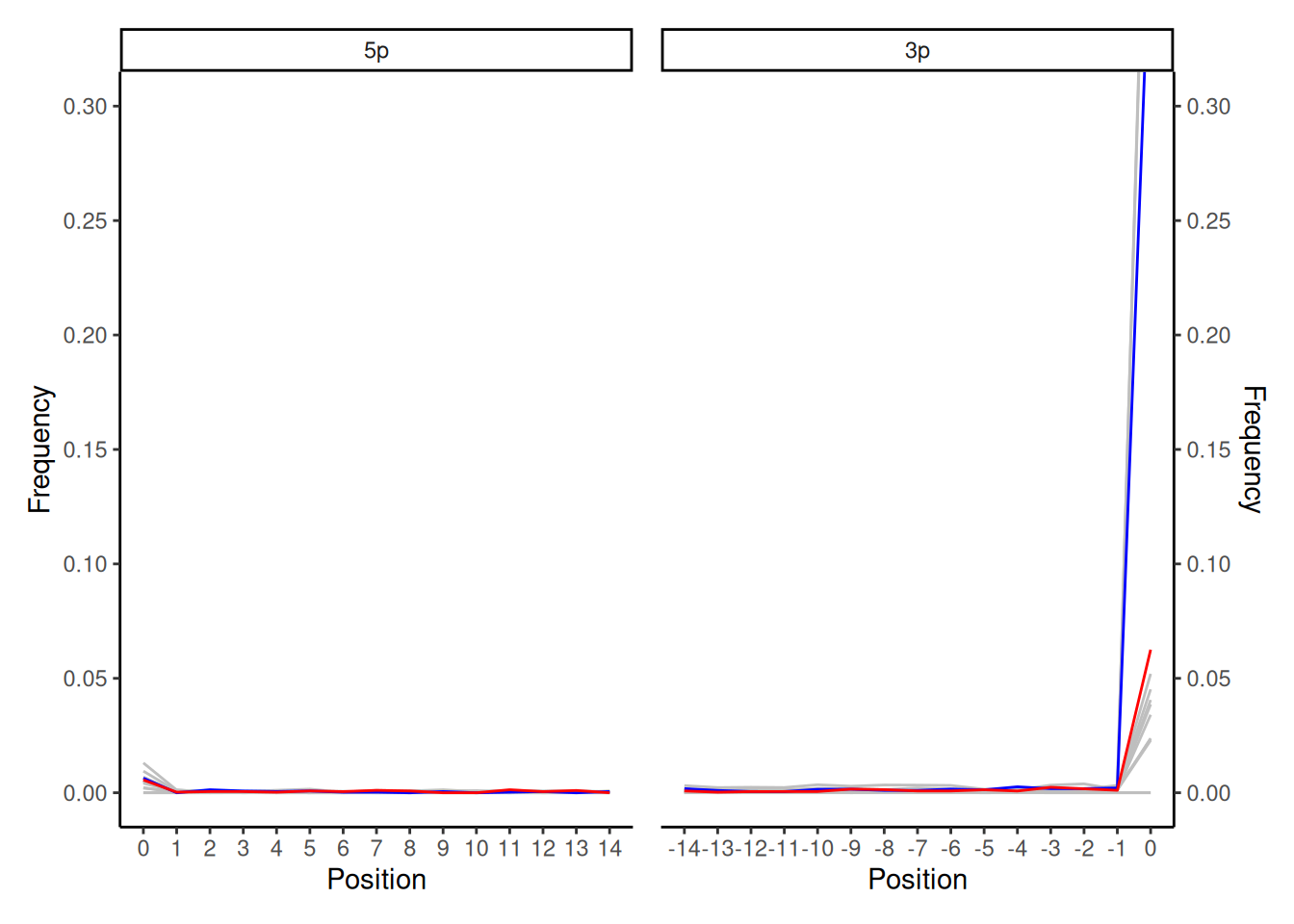
8 Left over adapter

This smiley plot was found from a paired-end sequenced, double-stranded, non-UDG treated library. It displays a very significant G to A frequency increase (exceeding 30%) on the first base of the 3’ read termini, as well as a small increase for C to T misincorporations and other base changes. This is likely due to a remaining base from an adapter that was not fully clipped off during the adapter trimming step of read preprocessing.
In the example above, the library was preprocessed with AdapterRemoval (v2.1.7) for adapter trimming and paired-end merging based on sequence overlap. With paired end data where the DNA ‘template’ is shorter than the number of sequencing cycles (read length), AdapterRemoval first aligns the two reads to each other. It then uses the information from the assumed ‘symmetrical overhang’ from the alignment, to identify and clip the adapters. In other words, the sequence of the template should exactly match, and thus the rest of the read that does not overlap is assumed to be adapter sequence. A limitation of the tool is that asymmetric read pairs (e.g., reads with leading Ns or differing insert sizes that can ‘shift’ the template overlap) can result in incomplete adapter removal or loss of authentic fragment termini. In this example, a single 3’ terminal adapter base remained after adapter trimming due to asymmetric overlap of the templates, leading to an artificial inflation of the damage frequency.
In other tools, this can be cause of the adapter sequence was not correctly specified (e.g., one base left off).
In case you get a similar plot and you still have your raw FASTQ files (and you probably do), you can easily reprocess them with a cleaner approach — e.g. another tool such as fastp (Chen et al. 2018), Cutadapt (Martin 2011) with Flash (Magoč and Salzberg 2011) (or another preferred tool for trimming/merging).