Public Channels
- # 2022-summerschool-introtometagenomics
- # 2023-summerschool-introtometagenomics
- # 2024-acad-aedna-workshop
- # 2024-summerschool-introtometagenomics
- # amdirt-dev
- # analysis-comparison-challenge
- # analysis-reproducibility
- # ancient-metagenomics-labs
- # ancient-microbial-genomics
- # ancient-microbiomes
- # ancientmetagenomedir
- # ancientmetagenomedir-c14-extension
- # authentication-standards
- # benchmark-datasets
- # classifier-committee
- # datasharing
- # de-novo-assembly
- # dir-environmental
- # dir-host-metagenome
- # dir-single-genome
- # eaa-2024-rome
- # early-career-funding-opportunities
- # espaamñol
- # events
- # general
- # it-crowd
- # jobs
- # lab-community
- # lactobacillaceae-spaam4
- # little-book-smiley-plots
- # microbial-genomics
- # minas-environmental
- # minas-metadata-standards
- # minas-microbiome
- # minas-pathogen
- # no-stupid-questions
- # papers
- # random
- # sampling
- # scr-protocol
- # seda-dna
- # spaam-across-the-pond
- # spaam-bingo
- # spaam-blog
- # spaam-ethics
- # spaam-pets
- # spaam-turkish
- # spaam-tv
- # spaam2-open
- # spaam3-open
- # spaam4-open
- # spaam5-open
- # spaam5-organizers
- # spaamfic
- # spaamghetti
- # spaamtisch
- # wetlab_protocols
Private Channels
Direct Messages
Group Direct Messages

@James Fellows Yates has joined the channel



@Fabien Voisin has joined the channel

@Mikkel Winther Pedersen has joined the channel

@Meriam van Os has joined the channel
To kick off the introductions:
👋 I'm James - I'm one of the instructors of the workshop 🙂. I'm currently based in Leipzig Germany, where I'm shared as a post-doc between the Leibniz Institute for Natural Product Research and Infection Biology - Hans Knöll Institute, and the Max Planck Institute for Evolutionary Anthropology (Germans really like long institute names for some reason...)
I don't actually work on environmental DNA - in fact my original background wasn't in biology but rather archaeology - but from my PhD I've been applying and developing ancient metagenomics techniques to dental calculus from ancient humans, Neandertals, and Chimps/Gorillas.
However many of the concepts and methods overlap very much! So one component of my postdoc has been trying to improve the quality (and user experience) of running standard metagenomic pipelines through the nf-core initiative, and other aspects of bioinformatic analyses. Another component of my postdoc has been establishing and running the SPAAM summer school, some of the material you have already been sent as pre-reading for the workshop you will be attendingin a couple of weeks 🙂.


@Matt Campbell has joined the channel

Hi, I’m Jamie and I’m a senior lecturer in environmental genomics at the University of Adelaide. My background is in the natural sciences, having studied geology and zoology before undertaking a PhD in palaeoecology. It was during the latter stages of my PhD that I first picked up a pipette and tried to extract DNA from coprolites of the extinct moa :poop_party:, sparking an interest in ancient DNA that has now lasted 18 years! Much of my research now involves applying ancient DNA tools to study past biological change – from understanding the evolution and extinction of species, to reconstructing past interactions between species, and resolving the impacts of human settlement on insular ecosystems.

@Roberta Davidson has joined the channel
Hi all, I'm Robbi, based in Adelaide I have a background in genetics and I just finished my PhD at ACAD at the end of 2023. In my PhD I worked on a combination of human ancient DNA in South America and benchmarking 1240k capture methods for human ancient DNA. Since finishing I've been working on various things including ancient soil microbiome research and Australian Indigenous genomics.

@Siobhan Evans has joined the channel
Hi all, I am Siobhan, a PhD candidate (about to be a year in) based in Adelaide at ACAD. My project focuses on sedimentary ancient DNA from caves within Australia with a strong focus on DNA preservation in this cave environment. I am also doing some work with stick-nest rat middens, using them as a source of ancient DNA. I look forward to meeting everyone when they arrive in a few weeks.

@Shyamsundar Ravishankar has joined the channel

Hello everyone, I am Mikkel and the second instructor. I am an assistant professor at the Centre for Ancient Environmental Genomics, University of Copenhagen in Denmark. I like James do not come with a background in genetics nor bioinformatics but has trained as a physical geographer. Which is where I gained my interest in past human and environments interactions, and started specialising and developing methods for how ancient eDNA can be used to study this from both geological and archaeological sedimentary deposits. And here I still am after almost 15 years. I strive to find/develop the best and most correct ways to process and validate ancient DNA from environmental samples and then there is an urge to push the boundaries of what is capable in both time and type of deposit.

@Vishwadeep Rout has joined the channel


@Georgia Thomson-Laing has joined the channel
Hi everyone! I'm Meriam and I'm a PhD student based at the University of Otago, Dunedin, New Zealand. I have a background in Forensic Science (Netherlands), but after doing a Summer project in genetics in NZ, I was hooked. I worked as a lab tech for a while, but my interests took me to do a graduate diploma in archaeology at Otago, where I got introduced to archaeogenetics- this was perfect! My Masters focused on the identification of pathogens in human remains from the Pacific. While I could not verify any with confidence, I did enjoy all the challenges it brought me! The bulk of my PhD will focus on analysing metagenomic data from kurī (extinct Māori dog) palaeofaeces- hoping to look at human-animal-environment interactions, such as settlement patterns, adaptation, and health and disease. I am very excited to come to ACAD and meet everyone!
Hello all - I'm Elisa and I'm a first year PhD student based in the Institute for Marine and Antarctic Science at the University of Tasmania. My background is mainly in molecular and theoretical evolution, but in reality I just really love old things and how bioinformatics can used to understand them. My research is aiming to studying and optimizing bioinformatic pipelines for aDNA/sedaDNA research to isolate and verify rare (e.g. low abundance) taxa in marine sedimentary records of the Southern Ocean. I'm really looking forward to visiting the mainland for the first time, and get stuck in at the workshop. Cheers!
Hi all, I’m Vilma and I’m a postdoc at the Australian Centre for Ancient DNA, University of Adelaide. My background is in environmental microbial ecology, and I’ve always been interested in studying how microbes adapt to harsh environmental conditions. This interest led me to use ancient eDNA to explore how microbial communities respond to environmental changes over time. I’m looking forward to meeting everyone at the workshop!😊
Hi all, I am Shyam, a research student at the Australian Centre for Ancient DNA, University of Adelaide, working on selection during the LGM. My background is in bioinformatics. I am fairly new to the world of ancient and environmental DNA. I used to build bioinformatic pipelines for medical genomics before this. I’m looking forward to learning more about environmental DNA and the challenges it presents.

@Lucinda Duxbury has joined the channel
Hello, I'm Lucinda, I'm from Adelaide but just moved down to Tassie for my PhD on ancient DNA from polar marine sediments (and maybe ancient RNA if we can find it...). I want to get waaay better at bioinformatics over the next 3 ish years, this seems like the perfect place to start. Looking forward to coming home, catching up, and learning lots!
Hi again.... wondering if anyone might be able to help with some pre-reading troubleshooting. I'm trying to do the tutorials for chapter 12 and have been running into problems with some of the packages in the bioconda channel needing python 3.11 to work. I'm remoting in to the UTAS HPC server - which I see only has up to python 3.6.8. Does anyone know a workaround for this? Thanks in advance for the help :)
*Thread Reply:* If you're installing into a conda environment, then conda it should install the right version of python.
Which step are you stuck on?
*Thread Reply:* Hey James, I'm stuck on 'conda env create -f denovo-assembly.yml'. It keeps failing to solve the environment. I have sent a screenshot of error message I get in the main chat (can't figure out how to attach here sorry!). Here is also an extract of this rather unruly string further detailing the problems: 'Solve for environment specs The following packages are incompatible ├─ pin-1 is installable and it requires │ └─ python 3.11.** , which can be installed; └─ pydamage is not installable because it requires └─ matplotlib-base but there are no viable options ├─ matplotlib-base [2.1.2|2.2.3|2.2.4|2.2.5] would re quire │ └─ python >=2.7,<2.8.0a0 , which conflicts with an y installable versions previously reported; ├─ matplotlib-base [2.1.2|2.2.3|...|3.3.4] would requ ire │ └─ python >=3.6,<3.7.0a0 , which conflicts with an y installable versions previously reported; ├─ matplotlib-base [2.1.2|2.2.3|...|3.5.3] would requ ire │ └─ python >=3.7,<3.8.0a0 , which conflicts with an y installable versions previously reported; ├─ matplotlib-base [2.2.4|2.2.5|...|3.7.3] would requ ire │ └─ python >=3.8,<3.9.0a0 , which conflicts with an y installable versions previously reported; ├─ matplotlib-base [2.2.5|3.3.2|...|3.8.2] would requ ire │ └─ python >=3.9,<3.10.0a0 , which conflicts with a ny installable versions previously reported; ├─ matplotlib-base [3.4.3|3.5.0|...|3.8.2] would requ ire │ └─ python >=3.10,<3.11.0a0 , which conflicts with any installable versions previously reported; ├─ matplotlib-base [3.6.1|3.6.2|...|3.8.0] would requ ire │ └─ pillow >=6.2.0 but there are no viable options │ ├─ pillow [6.2.0|6.2.1] would require │ │ └─ python >=2.7,<2.8.0a0 , which conflicts w ith any installable versions previously reported; │ ├─ pillow [6.2.0|6.2.1|...|8.3.2] would require │ │ └─ python >=3.6,<3.7.0a0 , which conflicts w ith any installable versions previously reported; │ ├─ pillow [6.2.0|6.2.1|...|9.2.0] would require │ │ └─ python >=3.7,<3.8.0a0 , which conflicts w ith any installable versions previously reported;'
*Thread Reply:* Hey James, just for your reference, here is the entire error message I get: (base) [duxburyl@rosalind-01 denovo-assembly]$ conda env create -f denovo-assembly.yml Retrieving notices: ...working... done Channels:
- conda-forge
- bioconda
- defaults Platform: linux-64 Collecting package metadata (repodata.json): done Solving environment: failed
PackagesNotFoundError: The following packages are not available from current channels:
- conda-forge::visidata
- bioconda::samtools
- bioconda::pydamage
- bioconda::prokka
- bioconda::metabat2
- bioconda::megahit
- bioconda::maxbin2
- conda-forge::k8
- bioconda::gunc
- bioconda::fastp
- bioconda::diamond
- bioconda::concoct
- bioconda::checkm-genome
- bioconda::bowtie2
- bioconda::bioawk
Current channels:
- https://conda.anaconda.org/conda-forge/linux-64
- https://conda.anaconda.org/bioconda/linux-64
- https://repo.anaconda.com/pkgs/main/linux-64
- https://repo.anaconda.com/pkgs/r/linux-64
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/bioconda
- https://conda.anaconda.org/conda-forge
- https://conda.anaconda.org/conda-forge
To search for alternate channels that may provide the conda package you're looking for, navigate to
<https://anaconda.org>
and use the search bar at the top of the page.
*Thread Reply:* Ok for the time being can yuo open the environment.yml file and comment out (# at the beginning) or remove the conda-forge::k8s line
*Thread Reply:* That works for me
*Thread Reply:* I guess k8 is in thewrong place
*Thread Reply:* its been movd to bioconda
*Thread Reply:* So just replace conda-forge::k8s with bioconda::k8s
*Thread Reply:* Oh awesome - thanks James - I'll try that now 🙂
*Thread Reply:* Woohooo it's downloading now! I love coding again hahahah
*Thread Reply:* For future reference, is it a common occurrence that packages will switch between channels?
*Thread Reply:* Oh no.. for some reason the download just froze...
*Thread Reply:* > For future reference, is it a common occurrence that packages will switch between channels? No I'm a bit confused about it
*Thread Reply:* > Oh no.. for some reason the download just froze... Wht do you men?
*Thread Reply:* False alarm - turned out it was just me being impatient and I cancelled the command halfway through it running and then tried again and then that wasn't working (I think it must have been because some of the things were already downloaded and others weren't so it was getting confused or something) - so I reinstalled miniconda and started again and now it's working! Phew! 🙂
*Thread Reply:* Hello I'm back with another problem 😞
*Thread Reply:* Not sure why it isn't working but the last line seems to point towards python 2.7 being a little problematic (I feel like I shouldn't be surprised...)
*Thread Reply:* Hmm that worked for me... where did you run this command from? Were you in the newly created metamaps environment>?
*Thread Reply:* yes - i ran it from my home directory in the newly created metawrap-env
*Thread Reply:* not sure if relevant but just tried running from the /denovo-assembly directory and tried running it from my base environment - and neither of those worked either... hmmmm just another day and another problem running some code hey hahah
*Thread Reply:* Hmm I'm not sure sure then sorry... The issue looks to be there is an R version that it can't find but I don't know why it would be on yours and not my machine.
But anyway metawrap is not used in the stuff we are doing for the workshop, and I'm flying today so I won't be able to look... Therefore I suggest you just read the rest of the chapter and skip the commands that require netawrap output (iirc metawrap is one of the last sections anyway)
*Thread Reply:* Easy - no worries 🙂 Safe flight and thanks for all your help!

@Biancamaria Bonucci has joined the channel

For anyone else: it seems the k8 package moved from conda-forge to bioconda or something.
I've updated the Zenodo record with the correct/working environment.yml file.
@Lucinda Duxbury you can either manually edit the file as I mentioned, or download the Zenodo again (the same DOI/link on the summer school page should work 🙂 )
Hi all, I had a question in regards to the prelim ahead of the workshop. Would it be best to run through the associated tutorials with the chapters, or just focus on the theory?
Chapter 6 is good to do if you're not particularly comfortable with working in the terminal.
For the rest it's fine to just read - they mostly are 'manual' versions of the pipeline sessions
(at least for the screening and assembly sessions)
Hi everyone!! I am Bianca and I am a PhD student based in Tartu (Estonia). My main project focuses on ancient dental calculus from Europe, spanning different time periods. I also perform wet-lab protocol development in my lab! Lately I have started working on soil samples from Neolithic Italy and Anglo-Saxon England. Looking forward to meeting you all in few days :smilingfacewith3hearts::catjam:

Hi all, Matt here and I’m a postdoc from the Trace and Environmental DNA Laboratory (TrEnD Lab) at Curtin University, Western Australia. I am currently investigating the potential of ancient sedimentary DNA as a tool for reconstructing Southwest Australia's paleoecology and paleoclimate during the late Quaternary period. For my Ph.D., I studied modern microbial mat communities using metatranscriptomics and organic geochemical techniques. Post-Ph.D., I joined the TrEnD Lab as a research technician facilitating laboratory operations and working on a variety of eDNA projects. In 2023, I become a post-doctoral researcher under the supervision of Prof Morten E. Allentoft.
Slides from my stuff for the whole workshop (@Mikkel Winther Pedersen’s will come later): https://drive.google.com/drive/folders/1bao_U6fi7eAHo3rwqIusVTxDOMBSXKVY?usp=sharing
And good morning everyone 👋
@channel username and passwords:
ssh ${USERNAME}@acadworkshop.uoa.cloud

mkdir bin && cd bin/
wget <https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh>

@Dawn lewis has joined the channel

@Kieren Mitchell has joined the channel
source ~/.bashrc
conda config --set auto_activate_base false
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge

sinfo -o "%P %c %m %t %l"
nextflow run nf-core/eager \ -r 2.5.0 \ -c envadna.conf \ -profile singularity \ --input '/shared/data/samplesheets/eagerinput.tsv' \ --outdir 'eager-screening/' \ --mapper 'bowtie2' \ --fasta '/shared/data/genomes/GCF000819615.1ViralProj14015genomic.fna' \ --runbamfiltering \ --bamunmappedtype fastq \ --metagenomiccomplexityfilter \ --runmetagenomicscreening \ --metagenomictool 'malt' \ --database '/shared/data/databases/maltdb/' \ --runmaltextract \ --maltextracttaxonlist '/shared/data/databases/maixnerminitargetlist.txt' \ --maltextractncbifiles '/shared/data/databases/' \ --skippreseq \ --skipdeduplication \ --skipdamagecalculation \ --skip_qualimap
nextflow run nf-core/eager \
-r 2.5.0 \
-c envadna.conf \
-profile singularity \
--input '/shared/data/samplesheets/eager_input.tsv' \
--outdir 'eager-screening/' \
--mapper 'bowtie2' \
--fasta '/shared/data/genomes/GCF_000819615.1_ViralProj14015_genomic.fna' \
--run_bam_filtering \
--bam_unmapped_type fastq \
--metagenomic_complexity_filter \
--run_metagenomic_screening \
--metagenomic_tool 'malt' \
--database '/shared/data/databases/malt_db/' \
--run_maltextract \
--maltextract_taxon_list '/shared/data/databases/maixner_mini_targetlist.txt' \
--maltextract_ncbifiles '/shared/data/databases/' \
--skip_preseq \
--skip_deduplication \
--skip_damage_calculation \
--skip_qualimap
Anyone else at smoking ceremony
Yes Bianca and I are in the middle standing
<#C05SQA7RA81|classifier-committee>
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 40G -t 01:00:00 --pty bash
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 10G -t 01:00:00 --pty bash
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 30G -t 02:00:00 --pty bash
conda install -c conda-forge -c bioconda -c genomewalker bam-filter
conda install -c conda-forge -c bioconda -c genomewalker bam-filter
srun -n 1 --mem 1GB vsearch --fastx_uniques euks_data/ERR10493277_small-FINAL.fq.gz --fastqout ./ERR10493277_small-FINAL.vs.fq --minseqlength 30 --strand both
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 10G -t 02:00:00 --pty bash
WHEN YOU'RE READY TO RUN VSEARCH RUN:
srun -n 1 --mem 1GB vsearch --fastx_uniques ERR10493277_small-FINAL.fq.gz --fastqout ./ERR10493277_small-FINAL.vs.fq --minseqlength 30 --strand both
srun -n 1 --mem 5GB sga preprocess -m 30 --dust-threshold=1 ERR10493277_small-FINAL.vs.fq -o ERR10493277_small-FINAL.vs.d1.fq
https://pubmed.ncbi.nlm.nih.gov/16796549/

for file in ERR10493277_small-FINAL.vs.d1.fq; do grep 'M_A00706' $file | cut -f2 -d@ > $file.readID.tmp ; done
grep 'M_A00706' ERR10493277_small-FINAL.vs.fq | grep -f $file.readID.tmp -v | cut -f2 -d@ > $file.readID_lowcom.tmp
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 10G -t 02:00:00 --pty bash
srun --export=ALL --ntasks-per-node 4 --nodes 1 --mem 10G -t 02:00:00 --pty bash
packages_to_install <- c("dplyr", "readr", "tidyr")
sbatch -n 1 --mem 8GB /shared/data/euks_programmes/readlengthPLOT_fastq.sh
keyboard cat
i dont know about you https://www.youtube.com/watch?v=AgFeZr5ptV8

and it looks like this:
```#!/bin/bash
SBATCH --nodes=1
SBATCH --time=2:00:00
SBATCH --job-name=bowtie2_Bianca
SBATCH --mem=30G
SBATCH --output=bowtie2.out
bowtie2 --threads 4 -k 1000 -x /shared/data/euksdatabase/refseq211smalldedup.fa -D 15 -R 2 -N 0 -L 21 -i S,1,1.15 -U ERR10493277small-FINAL.vs.d1.fq --no-unal | samtools view -bS - > ERR10493277small-FINAL.vs.d1.fq.refseq211small_dedup.bam```
and then i run it like this:
sbatch bowtie2.sh
https://github.com/biancamariabonucci/acad_workshop2024/blob/main/README.md
 <a href="https://github.com/biancamariabonucci/acad_workshop2024">biancamariabonucci/acad_workshop2024</a>
<a href="https://github.com/biancamariabonucci/acad_workshop2024">biancamariabonucci/acad_workshop2024</a>
@Biancamaria Bonucci, this is very good work on your repo! well done!! 🙌
*Thread Reply:* Thank you Mikkel, I highly appreciate it!
hey team, was thinking of going to see a circus show at the garden of unearthly delights tomorrow night (right by uni), if anyone else was keen, i was thinking of this one maybe which is like circus/comedy: https://adelaidefringe.com.au/fringetix/yuck-circus-af2024 at 7pm

*Thread Reply:* This seems super nice, count me in!

nextflow run nf-core/mag \
-r 2.5.3 \
-c envadna.conf \
--max_memory 30.GB \
-profile singularity \
--input '/shared/data/samplesheets/mag_input.csv' \
--outdir ./mag-assembly \
--skip_spades \
--skip_spadeshybrid \
--ancient_dna \
--binning_map_mode own \
--skip_concoct \
--gtdb_db '/shared/data/databases/release214/' \
--busco_db '/shared/data/databases/bacteria_odb10/' \
--skip_krona \
--skip_metaeuk
Antonio's repos are really neat and he has designed the bam-filter tool among others. https://github.com/genomewalker His focus is on ancient microbes.
we have applied some of them in this preprint. but within the next 6 months we will release a ancient DNA tool kit (with loads of tools for ancient microbes). https://www.biorxiv.org/content/10.1101/2023.06.10.544454v1
Example mag output from the command today: https://www.dropbox.com/scl/fi/eatdtpgnu6f43b78l786q/mag-assembly.zip?rlkey=1epuxd0zgqus2hvesquhvs4fx&dl=0
Quick loop to check your input files are in the correct place…
for file in $(cat eager_prepared_input.tsv | cut -d$'\t' -f9 | tail -n+2); do ll $file ; done
nextflow run nf-core/eager -r 2.5.0 -c envadna.conf -profile singularity --input 'eager_prepared_input.tsv' --outdir ./eager-mapping --fasta '/shared/data/genomes/GCF_020735445.1_ASM2073544v1_genomic.fna.gz' --skip_fastqc --skip_adapterremoval --run_bedtools_coverage --anno_file '/shared/data/genomes/GCF_020735445.1_ASM2073544v1_genomic.gff.gz' --run_genotying --genotyping_tool 'ug' --run_bcftools_stats
nextflow run nf-core/eager \
-r 2.5.0 \
-c envadna.conf \
-profile singularity \
--input '/shared/data/samplesheets/eager_prepared_input.tsv' \
--outdir 'eager-mapping/' \
--fasta '/shared/data/genomes/GCF_020735445.1_ASM2073544v1_genomic.fna.gz' \
--skip_fastqc \
--skip_adapterremoval \
--run_bedtools_coverage \
--anno_file '/shared/data/genomes/GCF_020735445.1_ASM2073544v1_genomic.gff.gz' \
--bam_unmapped_type discard \
--run_genotyping \
--genotyping_tool ug \
--gatk_ug_out_mode 'EMIT_ALL_SITES' \
--gatk_ug_genotype_model SNP \
--run_bcftools_stats
https://x.com/willovvens/status/1760400254065947047?s=46&t=dn0VpPNJ3uT7U-FbUXatPA


^ for the 6 people who confirmed! https://adelaidefringe.com.au/fringetix/yuck-circus-af2024 <- follow this link if others are keen
Random question: does anyone want a half-dozen eggs 😅?
Hey all - we're planning Semaphore Beach during the free afternoon today. The current plan is to leave from campus around 4 pm, so that way people could go fetch some swimmers of the like! Also, would anyone local be keen to go and could carpool? Lucinda has her car in today, but probs wont be able to fit everyone in the car

Eggs: yes And sorry I’m busy this afternoon and can’t help with carpooling
*Thread Reply:* @Lucinda Duxbury also volunteered, I will just bring them tomorrow morning and you can decicde who wants them more 😆
*Thread Reply:* i’ll fight her for them… but she’s scrappy so I suspect she’ll win
*Thread Reply:* Could do an quick-fire exam and whoever gets the most number of right answers gets the eggs 😬
*Thread Reply:* (jk I hate exams)
*Thread Reply:* I am technically a volunteer fire fighter so might be in with a fighting chance

Super late introduction, sorry team! 😅
I'm Thom, a budding MPhil student (hopefully converting to PhD in about a year) under Jamie at ACAD here at the University of Adelaide, and I'll be looking at ancient DNA to analyse what species were present in sediment and cave samples from across the country, with the hope down the line for the information to go into a searchable database for species reintroduction efforts undertaken by groups such as NPWS, WWF and Bush Heritage. I'm originally from Aotearoa New Zealand :flag_nz:, where I did my Microbiology undergrad at the University of Otago, and I did my Master of Conservation Biology at Macquarie University in Sydney. I moved to Adelaide roughly August last year (time has really flown!), and it's been great meeting everyone over the course of the workshops so far this week!
If you REALLY want to look into the SAM spec: here are all teh tags
The full specification: http://samtools.github.io/hts-specs/SAMv1.pdf
srun --export=ALL --ntasks-per-node 1 --nodes 1 --mem 1G -t 02:00:00 --pty bash
source /apps/software/functions.sh
/apps/software/metaDMG-cpp/misc/compressbam --input [BAM] --output [OUTPUT_BAM]
diff -y <(samtools view -H ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L21.N1.bam) <(samtools view -H ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L21.N1.comp.bam)
conda env create -f /shared/data/euks_env/bam-filterENV.yaml
conda activate bam-filter3
pip install bam-filter
Figure 1 has a nice visualization of the LCA

filterBAM filter -e 0.6 -m 8G -t 4 -n 3 -A 92 -a 95 -N --bam ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.bam --stats ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.bam.stats.tsv.gz --stats-filtered ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.bam.stats-filtered.tsv.gz --bam-filtered ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.filtered.bam
zcat ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.bam.stats.tsv.gz | tabview -
Hey everybody! Tomorrow’s dinner will be in Pizza e Mozzarella (33b Pirie Street) Bar from 6:00 PM to 7:45 PM. We’ll have a chef selection of pizzas and lettuce salads 🙂 https://pizzaemozzarellabar.com/












*Thread Reply:* wait, is fasta pasta an adelaide thing?
*Thread Reply:* pasta is actually just an acronym for pastAncient
*Thread Reply:* > wait, is fasta pasta an adelaide thing? > No clue, but I've seen a couple of them here now...
Your food also gets delivered by a cat here.



Umm @Elisa Davis... your gargammel problem might already be solved, had an epiphany in the shower: https://bioconda.github.io/recipes/gargammel-slim/README.html
Check the description!
A new and relevant paper for your morning coffee https://onlinelibrary.wiley.com/doi/10.1002/edn3.514
And @Roberta Davidson wanna make your own metromaps wiothout having to learn graphic design? Rike posted this on the nf-core slack: https://tennessine.co.uk/metro/
*Thread Reply:* Yessss!! I was actually just thinking of making one for a paper yesterday
*Thread Reply:* DO IT
If anyone wants to try some Indigenous Australian foods, there is a stall in the central markets which is closed Sundays but open today until 9pm tonight. https://shop.adelaidecentralmarket.com.au/collections/something-wild
Who is keen to go on a walk tomorrow at Horsnell Gully? We’re sorting out car pooling https://www.parks.sa.gov.au/parks/horsnell-gully-giles-conservation-parks

*Thread Reply:* I am keen!
*Thread Reply:* Which time did you want to leave?
*Thread Reply:* that's a very Dawn-like reply 🤣
https://github.com/genomewalker/bam-filter
Try this for viewing the table :):
zcat ERR10493277_small-FINAL.vs.d1.fq.refseq211_small_dedup.L22.comp.reassign.bam.stats-filtered.tsv.gz | column -t -s $'\t' | less -S -N

@Elisa Davis the data is here /shared/data/euksdata/ERR10493277small-FINAL.vs.d1.fq.refseq211smalldedup.L22.comp.reassign.bam.stats.tsv.gz

*Thread Reply:* We aren't sure exactly what this means. The references with 0 coverage evenness are probably bad though...
Breadth vs Exp Breadth to show the eveness of cov for each contig

Exp breadth vs evenness of coverage with the reference length shown by colour gradient


Plot cov_eveness vs exp breadth filtered and unfiltered


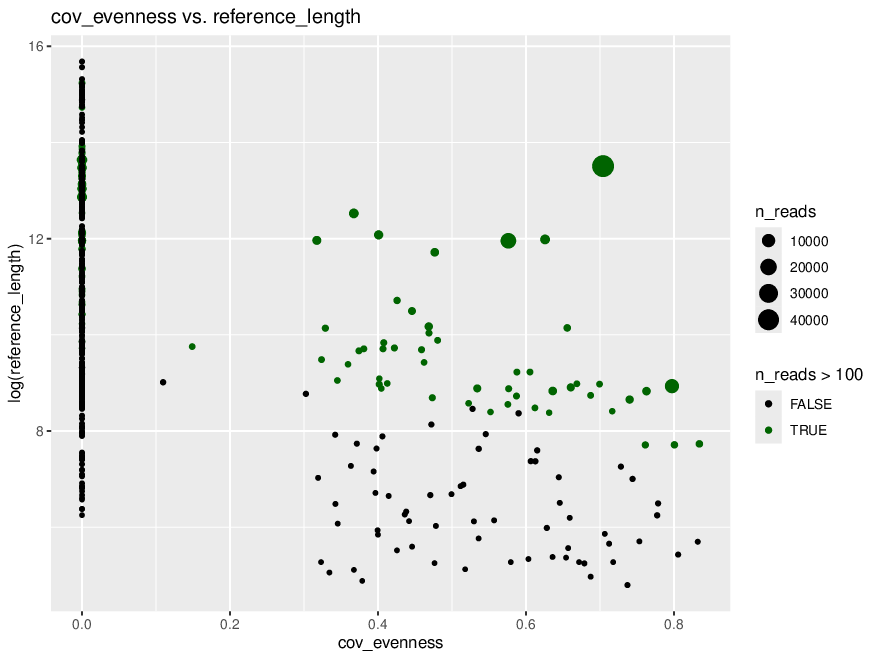

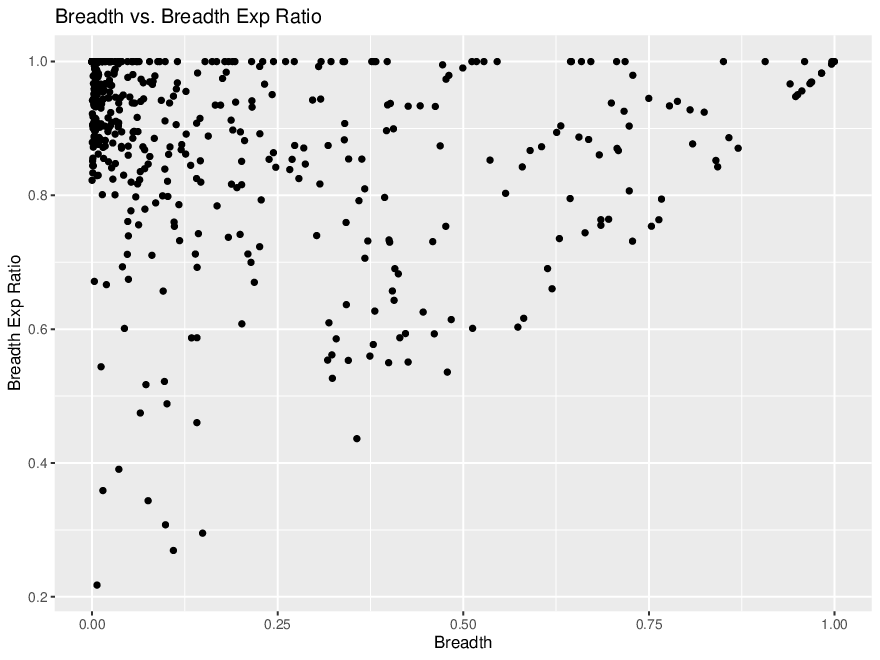


The top left have low cov_eveness indicating stacking of reads in maybe conserved regions.

srun --export=ALL --ntasks-per-node 3 --nodes 1 --mem 8G -t 02:00:00 --pty bash
source /apps/software/functions.sh
metaDMG-cpp lca --names /shared/data/euks_taxonomy/names.dmp --nodes /shared/data/euks_taxonomy/nodes.dmp --acc2tax /shared/data/euks_taxonomy/small_accession2taxid.txt.gz --sim_score_low 0.93 --sim_score_high 1.0 --how_many 30 --weight_type 1 --fix_ncbi 0 --threads 4 --bam ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.bam --out_prefix ERR10493277_small-FINAL.vs.ds1.fq.refseq211_small_dedup.L22.comp.reassign.filtered
metaDMG-cpp dfit ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.bdamage.gz --names /shared/data/euks_taxonomy/names.dmp --nodes /shared/data/euks_taxonomy/nodes.dmp --showfits 2 --lib ds --out ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered
Only use this metaDMG-cpp version and repo https://github.com/metaDMG-dev/metaDMG-cpp
metaDMG-cpp aggregate ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.bdamage.gz -lcastat ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.stat.gz --names /shared/data/euks_taxonomy/names.dmp --nodes /shared/data/euks_taxonomy/nodes.dmp
zcat ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L21.N1.comp.reassign.filtered.bdamage.gz.stat.gz | paste - <(zcat ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L21.N1.comp.reassign.filtered.dfit.gz) > ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L21.N1.comp.reassign.filtered.combined_metaDMG_output.txt
zcat ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.bdamage.gz.stat.gz | paste - <(zcat ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.dfit.gz) > ERR10493277_small-FINAL.vs.fq.refseq211_small_dedup.L22.comp.reassign.filtered.combined_metaDMG_output.txt
Just adding the combined data for the fun this afternoon.


zcat ERR10493277small-FINAL.vs.fq.refseq211small_dedup.L22.comp.reassign.filtered.lca.gz | grep Mammut | awk '{print "> " $1 "\n" $2}'
Headers are fixed in this version (I think)
Thanks Kieren! the file is also here now /shared/data/euksdata/combinedmetaDMGout.txt.gz


```library(readr) library(ggplot2) library(tidyr) library(dplyr) library(stringr)
Set wd
setwd("C:/Users/duxburyl/OneDrive - University of Tasmania/PhD/Tassie PhD/Workshops and conferences/Bioinformatics workshop (Feb 2024)")
Load the table
data <- readr::readtsv("combinedmetaDMGout.txt")
Remove useless columns (using an magrittr R pipe cntl shft M)
dataforplot <- data %>% select("samplename", "name", contains("fwf")) %>% pivotlonger(namesto="position", valuesto = "frequency", cols=contains("fwf")) %>% mutate(position=str_replace(position, "fwf",""), position=as.numeric(position))
Make the plot
mycoolplot <- ggplot(dataforplot, aes(x=position, y=frequency)) + geompoint() + themebw()
ggsave("metadmgplot.pdf", device=pdf(), plot = mycoolplot)```
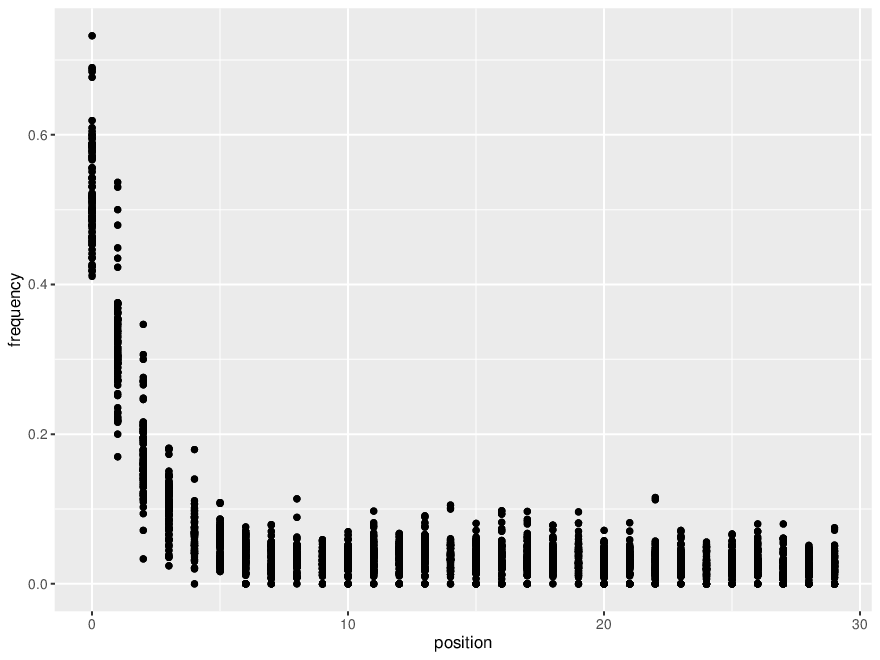
*Thread Reply:* ^filtered for plants


2000 colour palettes for R: https://emilhvitfeldt.github.io/r-color-palettes/discrete.html
I’m skipping dinner so my phone number is
Dinn er tonight: https://spaam-community.slack.com/archives/C06HATZKXFA/p1709176608580899
 }
}
Beers at MyLoverCindy on Pirie St - opens from 5:30pm
Proposal for tomorrow: • Sleep in followed by • casual brunch at the Adelaide Central Markets (oo la la) at 10:30am and THEN • a walk in the hills (Horsnell Gully - a well kept local secret you lucky bastards) with the option of either (time will be a limiting factor) • a cold one at the Scenic hotel or • coffee and cake at Ashton post office cafe
I have 3 spots in my car - my number is 0478 296 431
um only 1 spot remaining now......
CORRECTION: BEERS AT UNIBAR (Luci told me to say that)
I'm having some stomach issues so will not be joining dinner, but enjoy!
*Thread Reply:* Sorry to hear that, I hope you feel better soon!

*Thread Reply:* I love that there is absolutely no context for this - Thom we found your glasses!!!
*Thread Reply:* Ok that makes much more sense 🤣
*Thread Reply:* Thought you were just showing off your swag because why not 🤣
*Thread Reply:* look honestly por que no los dos
Hey guys!! For tomorrow Lucy and I are going tomeet around 9.30 at the central market, feel to join whenever ♥️
Dear all, a bit of SPA(A)M here!! Join the channel #spaamtisch to follow our series of seminars, as well as to come together with other nice people to tackle problems, speak about tools, methods, etc :spaamtisch: :spaamtisch:
Just confirming Liam and I only need to bring one car today?
i will probably be there closer to 9:45am sorry! add me on whatsapp
i do not have access to slack on my phone but can get whatsapp 🙂
From the tidy-master himself:
hadleywickham@fosstodon.org - Periodic reminder: The only way to write good code is to write tons of shitty code first. Feeling shame about bad code stops you from getting to good code.
https://fosstodon.org/@hadleywickham/112021309035884210






Hei guys!! Georgia and I are going to Semaphore today! Everyone welcome to join ☺️
Hey nerds, I made a start on the 'glossary of ancient metagenomics acronyms'. It's a markdown (.md) file that will go on the SPAAM GitHub when it's finished. (I think) you can fork it from my GitHub and add to it like that. Then I can approve those changes or something... I guess I will learn as I go! P.S. Does anyone know if you can sort table rows (e.g. alphabetically) in a markdown file?
<a href="https://github.com/lucindaduxbury/spaam-community.github.io">lucindaduxbury/spaam-community.github.io</a>
*Thread Reply:* sent pull request!!
*Thread Reply:* Or maybe you can make the little tutorial @Roberta Davidson 😏
*Thread Reply:* do i need to approve anything @Roberta Davidson - or can you access?
*Thread Reply:* That's the bit I'm not sure how to do
*Thread Reply:* I think you have to approve or merge or something
*Thread Reply:* @Lucinda Duxbury this is what you normally do when you get a pull request
*Thread Reply:* what is what you normally do?
*Thread Reply:* 1. Go to the pull request tab
- Click the one that you want to look at
- Read the opening comment on the default 'conversations ' tab to see what should be in the changes
- Click on the files tab to see all the proposed changes (red lines are original, green are the new changes/additions)
- If you have comments or want changes to the proposal you can press the green 'review' button in the top right, and leave the comment there - you can either leave a generic comment, an approval, or request changes (the latter means the PR can't be merged until the reviewer gives approval)
- Once you've given the approval, you go back to the conversations tab, and at the bottom you should see a 'merge' button
*Thread Reply:* And once it's merged, it's in your main branch and you can continuing editing but with Robbi's changes in it
*Thread Reply:* Oh sweet - thanks James! Robbi have you made any changes yet?
*Thread Reply:* Yes she has! Check the pull request tab on your fork :)
*Thread Reply:* ohhhhh i getit igetit igetit! 🙂
*Thread Reply:* codingcodingcoding!!!
*Thread Reply:* githubgithubgithub!!!
*Thread Reply:* Thanks James!!
*Thread Reply:* What is ‘Glossary of Ancient Metagenomic Acronyms’? Do you mean GAMA?
*Thread Reply:* I hear @Lucinda Duxbury screaming from here
*Thread Reply:* I hate that "GAMA" has a ring to it
It's the next SPAAM project! :mask_parrot:
Yes, it will go in the website when it's ready!
No you can't automatically order table rows unfortunately in basic markdown (there are IDE - hehehehe Plugins that can do it though probably)
@channel please let @Lucinda Duxbury of anything else missing (either acronyms, or definitions) and when Luci says she's ready I'll explain how to make a pull request from a fork and how reviews work :) can do a mini tutorial here if you want
A longer version, with screenshots is in the textbook:)
Albeit without the review section
So I had a dream about coding last night... I don't know if there is any going back from here?

In case anyone doesn’t have instagram / doesn’t follow acad (which you should!) here is the bit of footage from the end of the smoking ceremony
https://www.instagram.com/reel/C3035IJhmgT/?igsh=MXdsMWwxNGppNHZ4NQ==
@Shyamsundar Ravishankar @Roberta Davidson (or whoever at womad) in case you have slack on your phone - Budos Band, nice to listen to boring to watch so just stand under a mister where you can hear them 🤣
*Thread Reply:* Cool! I'm on +61401669032 if you wanna Whatsapp us
https://www.nature.com/articles/d41586-024-00520-y have you guys seen/heard about this?

*Thread Reply:* https://environmental-dna.ethz.ch/research/ercledna.html
Hello all! I hope you are all well!! A couple of things, first

your gift card has gotten prime location!!:)
second, I have been updating the https://github.com/miwipe/acad_workshop2024 so it should be up to date. Let me know if you have any issues at all!!?
third, Nicola Vogel, who created the Euka programme for fast mitochondrial identification of mammalian species has agreed to make a presentation of the tool and a tutorial. I need help to figure out when would be a good time during the day and then of course a date. All are welcome, also friends and colleagues.
fourth, we recently made a documentary about ancient DNA, is mostly about Eske and ancient DNA, but quite a few of the heavy aDNA PIs are featuring, and of course I am also there for my 15 sec of fame😂
you can find it here Hunt for the Oldest DNA [52” PBS NOVA], 2024 https://vimeo.com/904646278?share=copy PW: DNA2024
Hi all! I remember we talked about a tool that you can use to calculate how much more data you can generate from a library without exhausting it. Does anyone remember what it's called? 🙃
Metagenomics: Nonpareil