Public Channels
- # 2022-summerschool-introtometagenomics
- # 2023-summerschool-introtometagenomics
- # 2024-acad-aedna-workshop
- # 2024-summerschool-introtometagenomics
- # amdirt-dev
- # analysis-comparison-challenge
- # analysis-reproducibility
- # ancient-metagenomics-labs
- # ancient-microbial-genomics
- # ancient-microbiomes
- # ancientmetagenomedir
- # ancientmetagenomedir-c14-extension
- # authentication-standards
- # benchmark-datasets
- # classifier-committee
- # datasharing
- # de-novo-assembly
- # dir-environmental
- # dir-host-metagenome
- # dir-single-genome
- # eaa-2024-rome
- # early-career-funding-opportunities
- # espaamñol
- # events
- # general
- # it-crowd
- # jobs
- # lab-community
- # lactobacillaceae-spaam4
- # little-book-smiley-plots
- # microbial-genomics
- # minas-environmental
- # minas-metadata-standards
- # minas-microbiome
- # minas-pathogen
- # no-stupid-questions
- # papers
- # random
- # sampling
- # scr-protocol
- # seda-dna
- # spaam-across-the-pond
- # spaam-bingo
- # spaam-blog
- # spaam-ethics
- # spaam-pets
- # spaam-turkish
- # spaam-tv
- # spaam2-open
- # spaam3-open
- # spaam4-open
- # spaam5-open
- # spaam5-organizers
- # spaamfic
- # spaamghetti
- # spaamtisch
- # wetlab_protocols
Private Channels
Direct Messages
Group Direct Messages

@James Fellows Yates has joined the channel







@Meriam Guellil has joined the channel

@Nikolay Oskolkov has joined the channel


@Erkin Alacamli has joined the channel

@Ylenia Vassallo has joined the channel

@Chenyu Jin (Amend) has joined the channel

@Aleksandra Laura Pach has joined the channel

@Shahar Silverman has joined the channel


@Marwa A.Abdelazeem has joined the channel

@Biancamaria Bonucci has joined the channel

@Luisa Sacristan has joined the channel

@Cameron Ferguson has joined the channel
hey Thanks for your reminder @James Fellows Yates Shall I also generate a ssk key to connect with my terminal or shall I wait in this for our SPAAM course?
You won't need a key for the summer school 🙂
Good morning @James Fellows Yates 🌞 I joined gather right now . I am not that much familiar with gather, so I am just wondering where I have to enter 😊
Good morning! We will meet in about 50 minutes in the lecture room.
When you spawn in (should be by the 'front desk') follow the summer school arrows and then we will be in the big room on the right with loads of chairs
Textbook: https://www.spaam-community.org/intro-to-ancient-metagenomics-book/
@channel starting in 5 minutes
If you still have not received the VM email, your VM node URL is here: https://docs.google.com/spreadsheets/d/120pNr4uq_BVbRTWrqMs4heqtM8JuLgblzqNxIJCzWs0/edit?usp=sharing

Assuming you guys know this but thought I’d flag it anyway. (password for signing into the denbi node)

denbi is nt the most sophistricated


*Thread Reply:* You can click one ubuntu US/German
*Thread Reply:* Thanks @Chenyu Jin (Amend) ❤️

Sorry, where can I find the presentation that contains all the commands? I think I missed the link
@channel slight change of plan! The Python session will be in the room you entered whe nyou entered gather.town
So the lecture room or the lobby you spawn into?
@channel reminder that the structure of this year's summer school is a bit more group guided, so please talk to each other and help each other out! Have your webcam on if possible and go through the material together. You will learn better by hearing different perspectives etc, and different ways of explainning things (both explaning and lsitening!)











For reference: group three -

@channel starting in 2 minutes!

I might have missed if you mentioned @James Fellows Yates. Are the groups the same?
Yes groups same for the morning
R session extra exercsises: https://share.eva.mpg.de/index.php/s/rdSBnxxpkKyHDMB
R session extra exercises solutions: https://share.eva.mpg.de/index.php/s/BErm3qBmmaCid62
(you must use them in teh browser to download
To download with command line:
wget <https://share.eva.mpg.de/index.php/s/rdSBnxxpkKyHDMB/download/amd_exercise.R>
wget <https://share.eva.mpg.de/index.php/s/BErm3qBmmaCid62/download/amd_solution.R>

https://www.protocols.io/view/a-z-of-ancient-dna-protocols-for-shotgun-illumina-36wgq529xgk5/v2
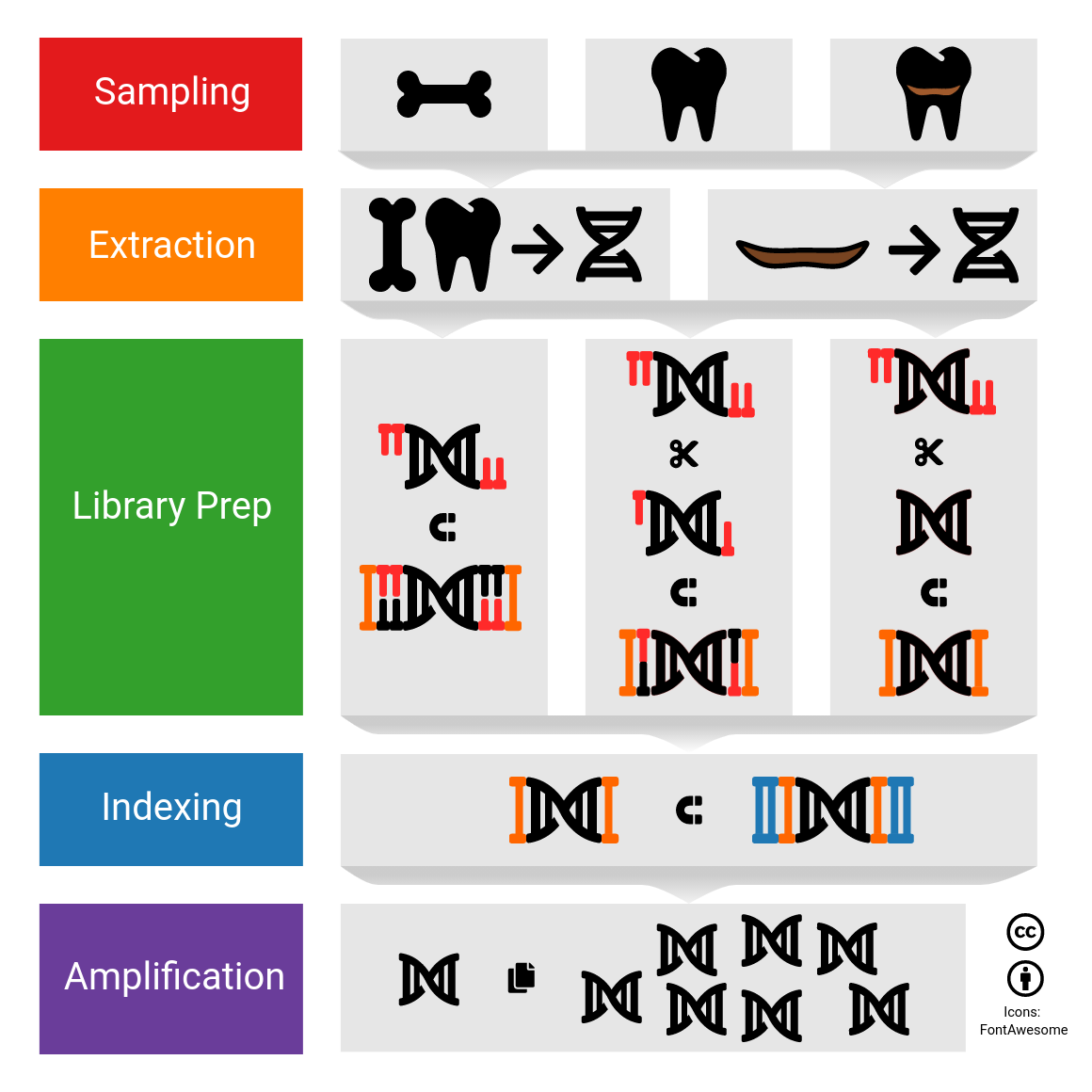
@channel my gather.town died catastrophically
So please ignore what I said, you're all free to go
We meet tomorrow at 9AM!
@channel
If when you spawn back today into the gather.town, and you're still on the beach/terrace area, please click on your name in the bottom right of the window and press 'respawn' . This will take you back to the main office/lobby room

the window appears in the bottom Left.
Dear @James Fellows Yates Can you share with us the criteria of yesterday filtering on AMDir viewer tool one by one till reach the files as I wasn't able to follow, Another question will we use theses files in the next practical session? Thanks in advance 🌸
Sure!
That is in this chapter of the text book (And this is one is working because I wrote this from scratch [other than the github]
But no, those files will not be used in a future session so don't worry about that
Many thanks @James Fellows Yates 🌸👍

Will you and Christina share the slides with us? Or is it already posted somewhere on SPAAM?
Yes all slides will be on the website after the summer school 🙂

Command to run when Maxime asks you to (⚠️ do not run beforehand!)
cd /vol/volume/taxonomic-profiling && rm -rf ** && git clone <https://github.com/maxibor/microbiome_tutorial.git>
Once glt clone has finished you can cd microbiome_tutorial/
By 'backup' I just mean move the files out of ~/Downloads
I will run rm -r ~/Downloads/** on allof your nodes
Deleting the contents of ~/Download/ in 1 minute!
Link to genome mapping: https://www.spaam-community.org/intro-to-ancient-metagenomics-book/genome-mapping.html

Hi! Since we're in the topic of genome mapping, I wanted to ask a technical question but it's not related specifically to the practical we're following. It's about the bowtie aligner. With ancient DNA, why is bowtie often used with the sensitivity flag and not end-to-end?
*Thread Reply:* You mean why people use local over end to end/
*Thread Reply:* sensitive applies to both mode

*Thread Reply:* https://www.frontiersin.org/articles/10.3389/fevo.2020.00105/full This might help though, if you've not already read it
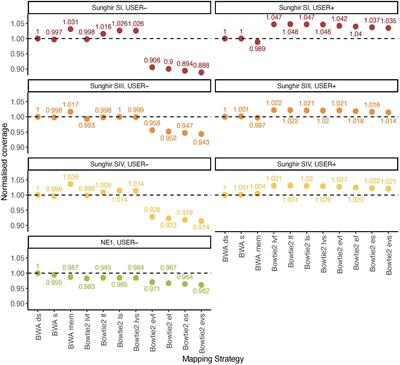
*Thread Reply:* Ah yes sorry, I meant local. Thank you for the paper!
*Thread Reply:* Note that our department are mostly BWA users (although the difference is quite small between the two)
*Thread Reply:* @Maxime Borry has some bowtie2 experience though
*Thread Reply:* @Rajiv Boscolo Agostini "end-to-end" is a global mode of bowtie2 alignment. In aDNA reserach, people prefer global alignment because it ensures the C/T polymorphisms are not soft-clipped as it is often the case for local (i.e. not "end-to-end") alignment. For the same reason people prefer "bwa aln" instead of more efficient "bwa mem", because the latter does local alignment which is not optimal for aDNA research. Basically, for aDNA applications, I would not run bowtie2 with local flag but only with end-to-end. Now, "sensitive", "very-sensitive", "fast" etc is not related (as far as I know, and as James said) to local-global choice but it is a special mode, i.e. a combination of low-level parameters which for simplicity was wrapped up under the "sensitive" flag
*Thread Reply:* Thanks @Nikolay Oskolkov!
*Thread Reply:* I wonder if you could write a short blog post on that @Nikolay Oskolkov? 😬 (@Ele and @Shreya)
Would it be possible to download the book as pdf in the future? Currently unavailable.
In the future yes, but currently not, book formatting is much more complicated than web 😬
After the summerschool after we get the few remaining corrections I will make a PDF version but it will be without proper formatting
I have a question about Friday: In the schedule it says, that the last time slot is reserved for a Dinner in Leipzig. When does it start? (I guess not at 15:45h, even though it says so in the time table)
I've not made a reservation yet! If you are in Leipzig reaching distance and would like to join please DM me !
You are also welcome to come to MPI-EVA for the whole day if you wish, we are in ateaching room so there is loads of space
(but normally we would go somewhere between 6 or 7

🫣Don't forget to share your dinner here that day🤣









3 tools (named after ghosts):
general plotting pool based on multiple bam files (plottergeist) one great binning tool (possibly also working with aDNA) (binshee) database subsampler for huge databases (which then runs a classifier on the smaller databases) (subspecter)

3 tools of group 2 (named based on Sherlock Holmes)
- Moriarty - searches through databases and finds sequences with adapters residues and nonsense DNA sequences and gets rid of them. No more wrong assignments to carps!
- Holmes - tracks changes of nomenclature of organisms for you. No need to remember what was that bacteria called 10 years ago!
- Watson - supports you on your taxonomic journey, will compare outputs of different tools for you and will find common assigments. More time for you and your important work on other analyses!
@channel Great job everyone! You had so many fantastic and creative ideas!
For those who asked me about dyslexia friendly font: https://opendyslexic.org/
How you configure in your Terminal will depend I'm the terminal, but most modern ones support it
It may reduce certain common errors by people with dyslexia a bit :)
Looking through these again, I the Plottergeist, Binshee and Subspecter are my favourites, they are still making me giggle now 😆 good job @Aleksandra Laura Pach and co!
unironically great names 😂
Good Morning Dr. @James Fellows Yates I opened My VM and IGV on the server a couple of minutes ago but it is not responding to any action. Any suggestions to solve this. Thanks in advance 🌸
Good morning! Let me have a look (and no need for the Dr. 😬 , makes me feel old... 😉 )
Thanks 😊 @James Fellows Yates Dr. Just for appreciation 😅

Hi , I think I have a problem with my Unbuntu. when I try to run the commands it appears that all of the programs like samtools and bowtie2 and the other programs that we used are not installed anymore. is there a way to fix this or do I need to install all of them?
I'll log on to your node, lpease wait a moment
You had somehow exited the conda enviroimnent 🙂
I've loaded it back for you
Reminder: Please don't download the simulated data!
Reminder: Please don't trim the reads!
Reminder: don't download the RefSeq database! Don't run Krakenuniq!
@channel about to start again!
(but if you want you can go partial...)
@channel remember, one quadrant per person!
(unless you have three people, in which case one person can have two)
@channel don't forget the name!





Group 6 (@Erin E Barnett, @Luisa Sacristan, @Aurore Galtier & me) 🤡☕

Group 7 Charlotte-deamination:


@Aleksandra Laura Pach I think it was this:
https://github.com/muellan/metacache
https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-3429-6

And an more fancy one: https://github.com/jmabuin/MetaCacheSpark

@channel please check now already if your deNBI node is still working etc!
Well done and thank you again to everyone for getting through the course! It is very intensive and really the course in the 'incubation' phase, so we thank you for your patience when we were going through the technical hiccups!
We hope the knoweledge you gained is useful ❤️
Dropping this here as it’s related to the text book for the course. I recently set up nf-core eager and was attempting to replicate the analysis in the text book as a test run however when I attempted to run the commands:
```## Download from NCBI curl -OJX GET "https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCF_001293415.1/download?include_annotation_type=GENOME_FASTA,GENOME_GFF,RNA_FASTA,CDS_FASTA,PROT_FASTA,SEQUENCE_REPORT&filename=GCF_001293415.1.zip" -H "Accept: application/zip"
unzip .zip mv ncbidataset/data/GCF001293415.1/ .
We have to sort the gff file to make it eager compatible
gffread genomic.gff GCF001293415.1ASM129341v1genomic.gff``
I got the following error:
gffread genomic.gff GCF001293415.1ASM129341v1genomic.gff
Error: cannot open input file GCF001293415.1ASM129341v1_genomic.gff!`
I thought it would just be a matter of specifically passing the -o flag before GCF001293415.1ASM129341v1_genomic.gff. This did resolve the error here but I got a different one when running the pipeline:
```Error executing process > 'bedtools (ERR4093961)'
Caused by:
Process bedtools (ERR4093961) terminated with an error exit status (1)
Command executed:
## Create genome file from bam header samtools view -H ERR4093961_rmdup.bam | grep '@SQ' | sed 's#@SQ SN:\|LN:##g' > genome.txt
## Run bedtools bedtools coverage -nonamecheck -g genome.txt -sorted -a GCF001293415.1ASM129341v1genomic.gff -b ERR4093961rmdup.bam | pigz -p 1 > "ERR4093961rmdup".breadth.gz bedtools coverage -nonamecheck -g genome.txt -sorted -a GCF001293415.1ASM129341v1genomic.gff -b ERR4093961rmdup.bam -mean | pigz -p 1 > "ERR4093961rmdup".depth.gz
Command exit status: 1
Command output: (empty)
Command error: Error: Sorted input specified, but the file GCF001293415.1ASM129341v1genomic.gff has the following record with a different sort order than the genomeFile genome.txt NZCP009972.1 RefSeq gene 504 740 . + . ID=gene-AK38RS24590;geneID=gene-AK38RS24590;genename=AK38RS24590
Work dir: /cluster/project7/AncientPathogen/testresults/work/33/fc706f7e45b8a0308d82741e5db37d
Tip: when you have fixed the problem you can continue the execution adding the option -resume to the run command line ```
Which seems to me like the reordering step didn’t work, but its hard to tell as the text book doesn’t really mention what exactly makes a gff file eager compatible. Any ideas what’s going on?
*Thread Reply:* Uhhh that's interesting
*Thread Reply:* Gffread is what worked for me... There are various tools that should be able to sort the gff file I saw when googling it, maybe you can check for an alternative until I have a chance to fix it.
I can also send the gff file I prepared before maybe later or tomorrow.
Currently have to juggle baby and Ikea furniture 😅
*Thread Reply:* If you could find time to send the gff file you prepared I'd appreciate it
*Thread Reply:* And I'll also have a look into other tools as well in the interim
*Thread Reply:* This should have the entire dataset stuff for the pipeline session
*Thread Reply:* The gff should be in there
*Thread Reply:* Note the fine is 4.8gb!
*Thread Reply:* Because it contains the conda envs for aMeta,
*Thread Reply:* I've not had a chance to clean that up
*Thread Reply:* OK I'll make a note to clean it up after download, thanks for your quick response and help
Btw I will be posting updates here for each session once I've cleaned everything up. I've almost finished doing the proper git/GitHub chapter
If anyone else would like one of the more problematic sessions fixed, I can make that a priority. Please let me know!
@channel I've finished and pushed a **completely revamped GitHub tutorial.
https://www.spaam-community.org/intro-to-ancient-metagenomics-book/git-github.html
You don't need any data to download and only need git, if you wish to try this again!

Hi! I am attempting to set up the functional profiling environment on my institutions cluster and I ran into this error. Any ideas how to fix it?

*Thread Reply:* I think it's because it's been moved to bioconductor: https://www.bioconductor.org/packages/release/bioc/html/decontam.html
*Thread Reply:* Which mean it's now served in Conda Land by the Bioconda recipe rather than conda-forge
*Thread Reply:* Soooo replace that line in your conda.yml with: - bioconda:: bioconductor-decontam
*Thread Reply:* (not tested, from my phone )
*Thread Reply:* If it works let me know @Erin E Barnett then I can update the environment
Hi all!
It was lovely meeting some of you at ISBA10!
I've finished revising the Accessing Metagenomic Data with AMDirT (plus git practise) chapter: https://www.spaam-community.org/intro-to-ancient-metagenomics-book/accessing-ancientmetagenomic-data.html
Thank u so much James ☺️ ☺️ Great to meet you all!
Next up will be python/pandas and taxonomic profiling
So I have a question about something that was touched on but I don’t think it was directly discussed at the summer school. How does Bam rescaling work and when should we use it? Logically I feel like it must be related to library preparation, as in if I had a none-UDG library I’d use it and I wouldn’t for a full UDG library, but if that’s the case how should I handle a half-UDG library? Thanks in advance and if you have any links to further reading I’d appreciate it 🙂
My understand is it should still theoretically work with half-UDG, but most people just trim the first and last base off of each read to remove damage from half-UDG, it's less 'manipulative' and 'guesstimate' which is what happens when rescaling

Or see 'methods' second: https://academic.oup.com/bioinformatics/article/29/13/1682/184965
Also if you do bam rescaling, you have to be aware that you may be introducing a reference bias to your mapped data. That’s why trimming the last few bases of half-UDG data will be my recommendation
Python pandas verified!
Note the one change that will happen after I've gone through them all is I will change all the data uploads to Zenodo (rather than dropbox) and the conda environment file within the archive 🙂
But in the meantime you can already review from chapters 6-9!
@Maxime Borry’s taxonomic profiling chapter has been reviewed and should be working!
@Alina Hiss and Alexander's genome mapping session is reviewed and working!
@Nikolay Oskolkov's authentication and decontamination chapter reviewed and live 👌 https://www.spaam-community.org/intro-to-ancient-metagenomics-book/authentication-decontamination.html
Phylogenomics from @Arthur Kocher and @aidanva is now reviewed and online 👍
https://www.spaam-community.org/intro-to-ancient-metagenomics-book/phylogenomics.html
(just two more to goooo, then I try to generate the PDF and make a permanent 'release' of this edition)
@Alex Hübner’s de novo assembly reviewed! https://www.spaam-community.org/intro-to-ancient-metagenomics-book/denovo-assembly.html
*Thread Reply:* Thank you, @James Fellows Yates!
And finnnnalllyyyyyy Ancient Metagneomic Pipelines by myself, Megan and Nikolay
I will put the data and conda envs in a stable place and finalise the release and then you should have a stable edition (albeit still in a beta 'draft' state!) of the book 🙂
OK! There we have it: http://www.spaam-community.org/intro-to-ancient-metagenomics-book/
Final spa(a)m message from me (he). This is the final cleanup and tweaked material for this year! Unfortunately a PDF version is still not yet possible, I'm having to fight with LaTeX but if I get that working I let you know!
Please let me know if you hit any problems, and otherwise I wish you good luck with your ancient metagenomic research!
Ok final final thing! As above, the book above is only a draft and currently only contains the practical material.
But now, if you wish to recap any of the theoretical concepts from the morning lectures from @Christina Warinner @Meriam Guellil , Alexander Herbig, or myself, you can see these on YouTube!
https://www.youtube.com/playlist?list=PL3olEEj2ioAWPqnOglXhtTaMLb-j6KQHr
Unfortunately due to technical issues the quality of the recordings are low (Sorry about that 😞 ). But the quality of the audio and the slides should be good enough to read - and the slides are still on the summer school website (not the book!).
Happy ancient metagenomicking!

*Thread Reply:* Also @Christina Warinner @Meriam Guellil let me know if you see any mistakes - I did the uploads and info filling over a few days in spare moments so it wasn't a consistent workflow 🙈
*Thread Reply:* Awesome!! Thanks for all the things you've done😆