Public Channels
- # 2022-summerschool-introtometagenomics
- # 2023-summerschool-introtometagenomics
- # 2024-acad-aedna-workshop
- # 2024-summerschool-introtometagenomics
- # amdirt-dev
- # analysis-comparison-challenge
- # analysis-reproducibility
- # ancient-metagenomics-labs
- # ancient-microbial-genomics
- # ancient-microbiomes
- # ancientmetagenomedir
- # ancientmetagenomedir-c14-extension
- # authentication-standards
- # benchmark-datasets
- # classifier-committee
- # datasharing
- # de-novo-assembly
- # dir-environmental
- # dir-host-metagenome
- # dir-single-genome
- # eaa-2024-rome
- # early-career-funding-opportunities
- # espaamñol
- # events
- # general
- # it-crowd
- # jobs
- # lab-community
- # lactobacillaceae-spaam4
- # little-book-smiley-plots
- # microbial-genomics
- # minas-environmental
- # minas-metadata-standards
- # minas-microbiome
- # minas-pathogen
- # no-stupid-questions
- # papers
- # random
- # sampling
- # scr-protocol
- # seda-dna
- # spaam-across-the-pond
- # spaam-bingo
- # spaam-blog
- # spaam-ethics
- # spaam-pets
- # spaam-turkish
- # spaam-tv
- # spaam2-open
- # spaam3-open
- # spaam4-open
- # spaam5-open
- # spaam5-organizers
- # spaamfic
- # spaamghetti
- # spaamtisch
- # wetlab_protocols
Private Channels
Direct Messages
Group Direct Messages

@James Fellows Yates has joined the channel




@Sterling Wright has joined the channel
@channel this will be the open channel for attendees to chat. You can start inviting people now if yo uwant. We will shut <#CPHECT30A|spaam2-open> once spaam3 happens

@Gunnar Neumann has joined the channel






@Nikolay Oskolkov has joined the channel


@Alexandre Gilardet has joined the channel










@Mohamed Sarhan has joined the channel

@Corrin Laposki has joined the channel






@Claudio Ottoni has joined the channel



@Katerina Guschanski has joined the channel

@Alexandra Rouillard has joined the channel


@Ophélie Lebrasseur has joined the channel

@Javier González Serrano has joined the channel

@Clio Der Sarkissian has joined the channel

@Martin Nathan has joined the channel

@Scarlett Zetter (she/her) has joined the channel


spam spam spam
https://www.youtube.com/watch?v=mBcY3W5WgNU

*Thread Reply:* LMFAO I just realised the description is:
"Sing along to ’Spam Song' with this official karaoke style Monty Python lyric video. "

@Maria Lopopolo has joined the channel
@channel 🌅☀️GOOD MORNING and GOOD DAY EVERYONE!!!☀️ 🌅 Looking forward to kicking off SPAAM3 today. See you all in an hour! (6:00 am PST and 15:00 CEST)

For quick reference here is the link and meeting ID and passcode for the Zoom meeting: https://us02web.zoom.us/j/82064835502?pwd=Q0NzVWM3cGtnRm9Vb2pyK3Vha00zdz09 Meeting ID: 820 6483 5502 Passcode: 831478
*Thread Reply:* and program is here: https://spaam-community.github.io/#/events/spaam3/programme

Hello friends!
Could someone please tell me which email address the links came from. I can’t find them 😞
*Thread Reply:* I can forward to you 🙂
*Thread Reply:* For quick reference here is the link and meeting ID and passcode for the Zoom meeting: https://us02web.zoom.us/j/82064835502?pwd=Q0NzVWM3cGtnRm9Vb2pyK3Vha00zdz09 Meeting ID: 820 6483 5502 Passcode: 831478
*Thread Reply:* Most emails sent by Kelly: blevinske1@gmail.com
*Thread Reply:* (and forwarded to your york account : ))
*Thread Reply:* Thanks Kelly and James! Sorry about my disorganisation 🙄

*Thread Reply:* #SPAAM3 you mean? or the bone trafficer?
*Thread Reply:* https://twitter.com/search?q=%23SPAAM3&src=typed_query&f=live
*Thread Reply:* ah, just saw you replied with the actual hashtag 😆
Hi Rita! Wonderful talk. My questions are: (1) What policies or training programs would you recommend that research groups implement to ensure respectful treatment of individuals? (2) What is your thought on how this discussion extends to early-human or non-human samples? We have many ethical guidelines for living animals, but how do we stand on ancient ones?
Hi Justin, thank you for you talk! My question is: (1) With the drive towards increased data sharing between researchers from many nationalities, how do you think non-US researchers can build ethical relationships with the communities whose ancestors they would like to use as comparative populations? What specific methods do you think US/local researchers can/should use to facilitate discussions?
My question to Justin: Do you happen to know of any perspectives that N. American indigenous groups (such as the Navajo) have more specifically on the study of remains that aren't directly skeletal or 'human-made' material, such as dental calculus or palaeofaeces? I ask because like you said developing these relationships are important, but maybe topics such as microbial research isn't taught as much at Schools in general etc. and so improving this would be important for communication (This question is partly inspired what I've heard about the work lead by Tina Warinner's in Mongolia, where they realised they had to do a lot of fundamental education because some of their collaborators just didn't exactly understand what they were collaborating on) /overly long quesiton, sorry
Question for Justin: You mentioned throughout your talk very important points concerning native american communities (history, racism, integration, colonialism, etc.), particularly in relation to our work. My question are:
- Have the communities you are interacting with been in contact to discuss these topics with other native american nations across the Americas?
- Have you thought about elaborating codes of Ethics, and sets of guidelines regarding how Native American communities should be integrated, as other native communities worlwide have done in relationship to academic research?
Question for Justin/all speakers/SPAAM folk: Should we consider putting an ethics tag on our individuals' sample list to verify that some discussions/attempts at ethical conduct have been made? As well as warning tags for individuals' samples that may require more ethical groundwork prior to being researched?
@Jessica Hider do you mind sharing the slies already here? Would like to have them open during the discussion 😉

Hi Jessica, I may have missed this, but how are you incentivizing filling out this form? In biomedical studies, funding agencies won't provide funds if an approved IRB doesn't exist, but there isn't an expectation for this sort of thing from other funding sources. Will it be required by the university before they release funds to the researcher?
@James Fellows Yates, yes, working on it!! 🙂
Slides!

@Justin Lund do you know of any centralised lists of such organisations/conferences anywhere?
CARE: https://www.gida-global.org/care
https://www.nature.com/articles/s41597-021-00892-0
Check our Stephanie Russo Carol's work... Heres one to get you started. https://prod.repository.oceanbestpractices.org/handle/11329/1507
@Jessica Hider Suggestion for 4.8: be a bit more active "could the study negatively impact individuals or groups including," Say "What groups/individuals would be affected"
Brilliant first session, thanks for the great talks and discussion
Betsy your point is super valid about communicating skills and method choice but ultimately its the data generated that can be the most contentious ? For example when the samples are in "fasta" format what does that mean for these communities , how can the information in this format be used? Communicating can be more challenging than doing the least amount of destruction
*Thread Reply:* A lot of grants require open access at the end of project - how will datasets then be utilised? And then from a commercial point are companies (ie ancestry companies) using this open data in some framework for profit?
@James Fellows Yates, that's a great edit! Thanks you! 🙂
I am happy to share the whole working form if anyone really wants to get into it as well 🙂. Thanks everyone! I also wanted to say@Justin Lund and @Rita M Austin, great talks!! :)
Betsy's poll request from the ethics session:

*Thread Reply:* How is ‘indigenous/ethically sensitive contexts’ defined? Should e.g. Siberian sites be considered as such?
*Thread Reply:* Depends on your opinion 😉 but from my knowledge, yes, could be
*Thread Reply:* That is the main question Betsy had, not necessarily as clearly defined in Europe/Eurasia
*Thread Reply:* Yeah, I don't know how we could define it in Europe/Eurasia because there is not much discussion around it. Also the concept of "indigenous" itself inside Europe is not easy to define because there has been so much migration through Europe over the time. Just a bunch of thoughts here on the subject. But I think European scientists (including me) should try to include more local communities and people from the culture in the research process, indigenous or not indigenous.
@Meriam Guellil very interesting, dis you test the --minimizer feature of Kraken2 (that should be equivalent to unique kmer count in KU) against KrakenUniq?
@Ian Light I may have missed this during your talk, but in your database reduction are you only using full genome sequences? What about those species/strains that might only have genomes at the scaffold/contig/etc level of assembly? Do you expect that this might lead to some taxonomic drop out?
@Ian Light
1) Have you compare your workflow against the database construction part of the SPARSE pipeline? It also uses ANI (but difficult to use outside of SPARSE itself) 2) Do you have an approach to check for 'mislabelled' genomes that may accidentally be picked as a representative from a cluster (if I understood correctly)?
Cool work @Ian Light, how do you compute distance between your sequences and wouldn't you do multiple sequence alignment of all the sequences for all the ~25 000 ref genomes?

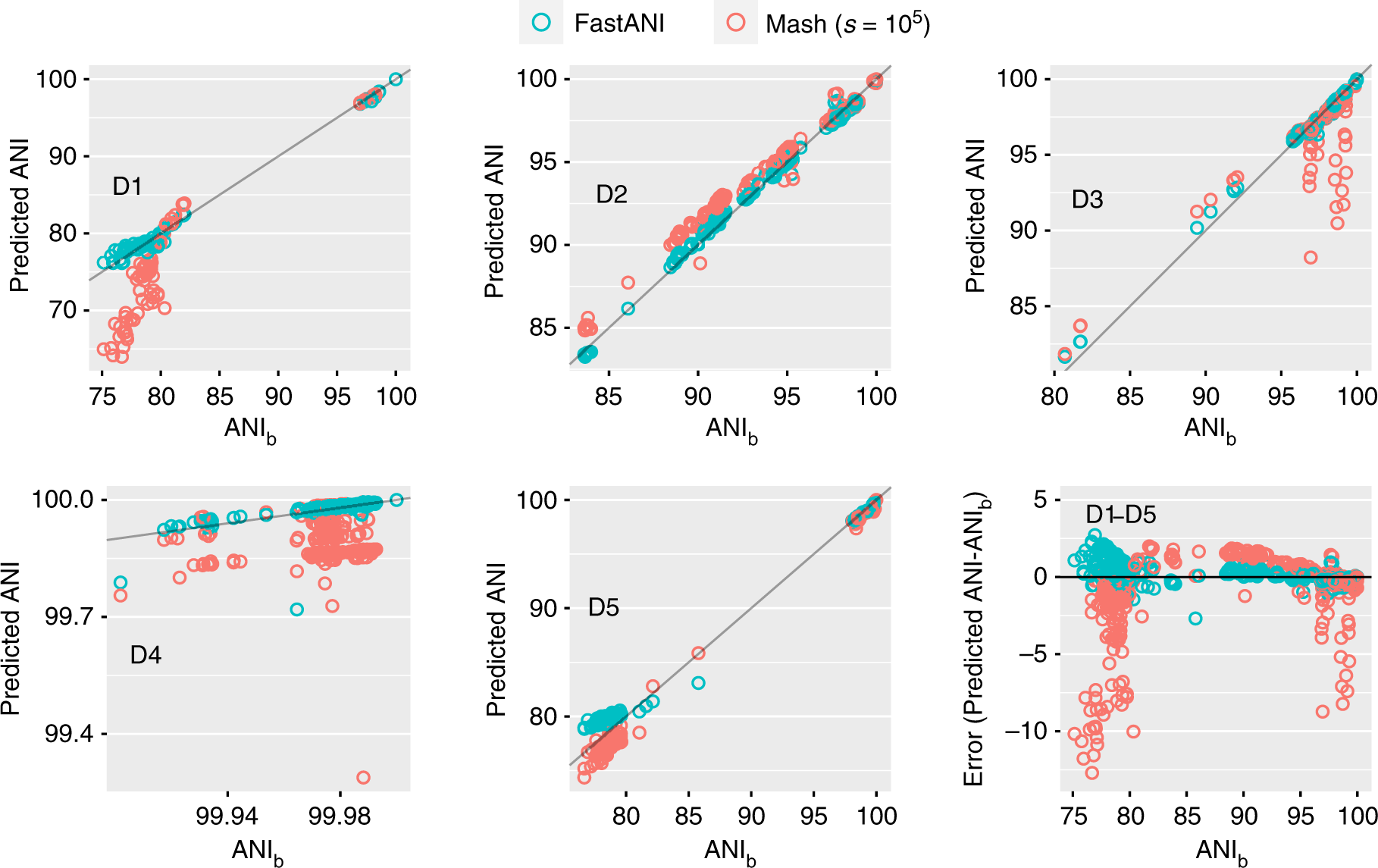
*Thread Reply:* we use fastANI for average nucl. identity calc. it is kmer based and impressively fast. https://www.nature.com/articles/s41467-018-07641-9
@Richell Ramírez Awesome project. I can’t wait to see what you recover! Do you know how the samples were selected? And how did you subsample the elements? Did you prioritize the lesions?
Paper Nikolay shared: https://pubmed.ncbi.nlm.nih.gov/28158639/

*Thread Reply:* Here is another great way to reduce redundancy in a database https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-018-0399-2


Hey @Ian Light, have you also taken a look into how much unique genetic diversity is lost by your reduction method?
(Add comments for other ones!)
Not specifically for pathogen detection but I’ve also used Diamond, Kaiju
Interested if krakenUniq outperforms malt (not speed but specificity/sensitivity)? anybody experience with both?
*Thread Reply:* @Felix Key I haven't seen a formal comparison but KrakenUniq allows basically an unlimited database size while MALT does not so I would expect KrakenUniq would be at least more sensitive, not sure about specificity though
*Thread Reply:* @Felix Key I did not compare MALT vs. KrakenUniq on synthetic data but ran MALT and KrakenUniq (using database built on same reference sequences) on a few benchmark samples where we are pretty sure we know the microbes. The results delivered by both tools were mostly in agreement, so no major discrepancy
Question to @Ian Light but also the whole audience: with more sophisticated approaches to build custom databases, have you thought about how to publish your methods guaranteeing full reproducibility?
*Thread Reply:* hi marcel we have not decided on that. guess a snakemake would be an option (and easy to rerun by other ppl) but not sure if that is what is going to happen at the end.
*Thread Reply:* Thanks, but do you think it would also make sense to publish the output (e.g. a file with all IDs of included refseqs)? Otherwise I would be afraid that screening pipelines of different labs are turning into ‘black boxes’ hampering comparability and also troubleshooting.
*Thread Reply:* sure databases grow constantly and interfere with explicit reproducibility. the approach ian presented is mainly a reproducible (non-random) method how genomes are selected and it should lead to a reasonable output whatever version of, lets say refseq, is used. of course for mirroring an exact database someone can always share the IDs used.
@channel reminder: please add your labs to: https://tinyurl.com/spaam3-labs
And also 🤫 <#C02D992D5TN|spaam-pets>

*Thread Reply:* every virtual conference makes me incredibly envious of all the amazing pets:meow_party:!! All i have our semi alive plants 🪴
*Thread Reply:* plants are great too!! I love a good jungle!!
@Kelly Blevins super interesting, wouldn't competitive mapping against all bacteria help fishing out truly MTBC reads? To my understanding, you use competitive mapping on a later stage (after you have mapped to MTBC ref genome alone) to discriminate between MTBC and other mycobacteria, but wouldn't it be informative to use competitive mapping as well at the beginning against the full database of all possible bacteria?

@Kelly Blevins Good job! With regard to the samples that do not have lesions, did you study the teeth of the individuals? . The interesting thing about this is that we study teeth because there are reports of treponemal DNA in teeth, so we analyzed the teeth to detect mycobacterium and we found the positive signal there.
@Kelly Blevins did you test the soil from the burial areas? With relatively low coverage I always worry that the mycobacteria I've detected are actually very closely related mycobacterium species--if you didn't test soil samples, do you have a way of verifying the hits are tuberculosis? Thanks!
Great talks all around!!
Where is the gather.town link?
SPAAM3_Islandvibe https://gather.town/invite?token=ZfpvIs5r SPAAMiscoolio2021
SPAAM3_Geekingout https://gather.town/invite?token=lyJnL4y0 SPAAMiscool2021

Or wsa it posted in zoom and I left too early 😱
Thanks a lot, this day was amazing!! Looking forward to to tomorrow’s talks (and @Petra Korlevic comics)
dude have patience!!
Cool kids go to the island
Can’t do gathertown today or risk further annoying labmates but will be there for tomorrow’s session. Have fun all! Drink a virtual beer for me 🍻
we needed two because we were more than 25 participants (that was the limit for free 😉)
Are you ok with having the discussion sessions included in the recordings?
Good DAY!!! #SPAAM3 day 2 is starting in 15 minutes!!! Join us in zoom for an exciting session on ancient metagenomics!!!
➡️ For quick reference here is the link and meeting ID and passcode for the Zoom meeting: https://us02web.zoom.us/j/82064835502?pwd=Q0NzVWM3cGtnRm9Vb2pyK3Vha00zdz09 ⬅️ Meeting ID: 820 6483 5502 Passcode: 831478
@channel! Remember to send your questions here!!
Hi @Abby Gancz how do your self-reported results about what metadata is collected compare to the metadata that is actually published?
And can you explain how 86% of people wear gloves in the lab? Is this actually 86% responded to this question? Or do you think people are really not wearing gloves?
(barking dog)
Thanks for your talk Abby! I'm new on the field so I would like to hear your main advice for best practices in ancient metagenomics. 🙂
(dog has been put in time out, so we can talk now in the future!)
@Abby Gancz thanks for the talk, is there any chance of standardising sampling procedures between archaeologists and biomolecular researchers when sampling?
osteology is very important
If @Abby Gancz asked our lab something we missed it b/c of the dog, hold on...
We have some preliminary work that points to more major differences, but I think it varies by populations
Marcel makes a good point! Field archaeologists and osteologists are collecting and not hearing these conversations. Might be an opportunity for a workshop??
We think so too! We're considering submitting one to AABA
*Thread Reply:* Yes please! (osteologist here 🙋♂️)
*Thread Reply:* What would be most useful to you in such a workshop?
*Thread Reply:* Oof, good question... lemme think about it
*Thread Reply:* I can also ask my lab what kind of things they would like to know
*Thread Reply:* @Abby Gancz I think the most effective sampling occurs in the field, right? So perhaps discussing the necessary equipment to wear while sampling (in an optimal scenario, but also considering practicality), as well as storage of the sample until sending it to the lab. Perhaps also a basic presentation of the potential contaminants and the effect they have on interpretation of the results.
Will the recorded talks be available somewhere? I missed the last session because of a meeting 😭
We will share the talks with the participants after the end of SPAAM3
The party room is doing SPAAM the correct way
https://www.protocols.io/view/dental-calculus-field-sampling-protocol-sabin-vers-bqecmtaw
@James Fellows Yates and @irinavelsko it can also be connected with OSF! 😁
*Thread Reply:* Yes... sort of... you can copy protocols directly to an OSF project
*Thread Reply:* I haven't explored it fully yet
*Thread Reply:* I know it's backed up on GitHub and clockss archived
*Thread Reply:* But this sounds awesome
*Thread Reply:* Where are the**

*Thread Reply:* Pretty well hidden and nothing in the docs (i think)
*Thread Reply:* OSF documented it: https://help.osf.io/hc/en-us/articles/360063021413-protocols-io
One such protocol @irinavelsko was referencing ☝️☝️ ☝️
@James Fellows Yates, you seem an ambassador for protocols.io 😉
(https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-6355-0 - written by Troll et al. No less)

*Thread Reply:* Yes, this is the method developed by our lab at UCSC. It works well for us
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8141684/
*Thread Reply:* @Nasreen Broomand is that what you referring to?
*Thread Reply:* we started using it but still a lot of room for improvement. I got many dimers
*Thread Reply:* @Maria Lopopolo try adjusting your iPCR cycles
*Thread Reply:* We're planning to start testing this method
*Thread Reply:* @Nasreen Broomand we do a qPCR before and we are adjusting it does help a bit but not so much. I am starting to think many times depends a lot from the quality of the DNA and also I notice smaller peaks like before dimers
*Thread Reply:* what is the beads to DNA ration you use to purify after amplification? we are using beads and not the elute kit
*Thread Reply:* @Maria Lopopolo there's another way to estimate cycle number without qPCR that I've found is easier and I get better results. I'll post that and the name of the cleanup enzyme I was talking about in a bit after the talks. We use 1.5x spri.
This is the method I was talking about:https://doi.org/10.1093/jhered/esab012
The restriction enzyme I was thinking of is XBA. iPCR cycles according to pmols: more than 1 pmol = 3-5 cycles 0.5-1 pmol = 4-7 cycles 0.25-0.5 pmol = 6-9 cycles 0.125-0.25 pmol = 9-11 less than 0.125= 10+ cycles I choose a number based on where my sample is within each range, and it works pretty well + no math 🙂
*Thread Reply:* What QC methods (e.g. qubit, tape, etc.) do you use to quantify and at which stage of your prep?
*Thread Reply:* @Nasreen Broomand thank you very much this is precious 🙂
*Thread Reply:* Hi Maria! We do a qubit reading in the ancient lab before library prep (in a special fume hood in a separate part of the lab since the qubit standard #2 obviously has a lot of DNA and we definitely don't want to contaminate anything!) Post-iPCR cleanup we just do qubit and tapestation 🙂
*Thread Reply:* our tapestation machine broke at one point so we were using a fragment analyzer for a while instead, which also works well. I wouldn't use nanodrop in lieu of qubit though if you're relying on those numbers for anything other than pooling
@Martin Nathan thank you very much for your very interesting talk! Did the bovine reads you detected from the sedDNA samples have damage pattern (I did not quite understand it)? And how would you explain the presence of elephant reads in your sedDNA samples?
*Thread Reply:* @Nikolay Oskolkov Yes we have been able to generate nice damage patterns for bovine reads. For the elephant, in Morocco it was an hypothesis that Loxodonta could have been present in Morocco and we detect it in 2 samples. So a bit surprising but not that much to retrieve some
@Allie Mann Cyprinos carpio was assembled on adapters, so your adapter trimming did not work
*Thread Reply:* It happens anyway
*Thread Reply:* The problem is that there are just so many adapters in the carp genome
*Thread Reply:* It picks up anything even if you still have like 5bp left of the adapter in there
*Thread Reply:* E.g. We adapter clip with even just 1bp overlap, and still get hits
*Thread Reply:* in my case cyprinos carpio signal always disappeared after I was more careful with adapter removal
*Thread Reply:* (sorry, pizza hands)
*Thread Reply:* Huh ok. What did you do instead?
*Thread Reply:* Or extra, rather
*Thread Reply:* It also picks up adapter artifacts whereby a stubby adapter ligates to an already-ligated stubby adapter
*Thread Reply:* two (or more) rounds of adapter trimming solves that
*Thread Reply:* Hmm interesting... Maybe something to implement in eager...
*Thread Reply:* @Pete Heintzman I do agree, we also run minimum 2 rounds of adapter trimming (and sometimes from different tools)
*Thread Reply:* would running adapter removal twice, remove bases that are actually not adapters? would that mess up with removal of duplicates?
*Thread Reply:* Had carp in a sample from Troy back in 2015 (as a new aDNAer). Told myself a whole story about how the person might have been buried with fish for some religious reason and was about to go do some research...mentioned it to a senior lab member and realized what was going on 🤣.
Question for @Allie Mann: When using environmental controls, would you suggest removing any eukaryotic species also detected in the environment?

@Allie Mann any thoughts on using a tool like krakenuniq to look at whether the # reads classified is just a result of very low complexity/highly conserved regions (few unique k-mers)?
*Thread Reply:* I actually was curious on how different our results would have been with krakenUniq — I started downloading the database last night but unfortunately didn’t get the analysis done before today
*Thread Reply:* i suspected as much 🥲: giving a talk knowing that someone will ask you "why didn't you do this thing that I too just learned that we should be doing"
@Allie Mann awesome talk! what would be the considerations when choosing the right database?
*Thread Reply:* I always try to use the full NCBI NT, basically all organisms ever sequenced by human being 🙂
my favourite was finding 🐼 and 🍍 in an early medieval Scandinavian sample once.
*Thread Reply:* something clearly had gone horribly wrong.
*Thread Reply:* I'm sure it's totally reasonable! They have been travellers, haven't they 🤪
*Thread Reply:* true @Katerina Guschanski perhaps I was sitting on the next Nature paper!! the bliss of being super green in data analysis 💚
*Thread Reply:* I once found a mammoth with malaria...
*Thread Reply:* Turned out it was PhiX contamination in a malarial genome.
*Thread Reply:* "mammoth with malaria" would make an impressive cover @Pete Heintzman - think of the media headlines :meow_party:
*Thread Reply:* I think my favorite was dolphin and cannabis in a mountain gorilla gut sample 😄
*Thread Reply:* Amazing!!! this is such a great thread for examples in teaching!
@Allie Mann Very interesting talk! I know there was one modern sample in the dataset. I was wondering if you were aware of any studies that include a larger dataset of modern calculus samples that also look at diet. This may sound strange as you can ask a living person what they are eating. However, it may us studying ancient datasets understand what and how much dietary DNA is recoverable? It may be more practical with populations with more traditional lifestyles.
@Nikolay Oskolkov gives me nightmares! Not that we never thought about it, just never so graphically!
@Nikolay Oskolkov On your story on contamination... i have experience the exact same... hits to bos taurus, the wild bore, but one of the hits i found was to Ovis canadensis... so when looking into it all reads mapped to one short contig... then blastN that contig, the highest hit was a pseudomonas...
@Nikolay Oskolkov we exactly have this problem with nice ancient Ursus sequences that revealed to be ancient microbes
if only I’d heard this talk before doing my first ever mapping! My results looked exactly like the picture with huuuuge spikes in some regions!

@Nikolay Oskolkov yes depth of coverage and coverage are often confused with each other. Sequence coverage is essential to detection
depth and breath of coverage should always be reported!!! (and breath at the depth you do your variant calling!!!)
Step-wise clasiffication, 1) with a specific database followed by 2) validation in a large database (such as whole NCBI nr db, with Blast when having just few thousands reads to validate) would be a good way to go when cluster capacity is limiting to load big DBs in RAM
*Thread Reply:* Actually this might be a good use of KrakenUniqs hierarchical database runs
@Nikolay Oskolkov wanna help add your workflow to eager? ;)
*Thread Reply:* We can discuss this James! We are going to publish this line of thinking soon (with Anders Göthreström and Love Dalen). Nf-core eager is definitely a good place to host this 🙂
But what Nikolay and Allison shown a very nice summary of all the limitations, cares and good practices that must be taken into account when analyzing ancient metagenomes. I think is good material for a very good review for a good journal if you are interested
*Thread Reply:* https://www.sciencedirect.com/science/article/pii/S1040618220307746?dgcid=rsssdall

*Thread Reply:* Yes! of course there is this one!
*Thread Reply:* but I got the feeling that some of the things mentioned have not been fully covered in that article.
*Thread Reply:* (Allie's figures were from that, but Indeed Nikolay had more in-depth examples Indeed)
*Thread Reply:* Also in this review: https://www.annualreviews.org/doi/abs/10.1146/annurev-micro-090817-062436 we also touch a bit, but it will be great to have a more specific publication
*Thread Reply:* It was while hearing to Nikolay that I thought about it
*Thread Reply:* Overall, I think it would make sense if there is enough material that hasn’t been mentioned in previous publications
*Thread Reply:* Thoughts @Nikolay Oskolkov and @Allie Mann?
*Thread Reply:* I would be happy to contribute!
*Thread Reply:* Me as well!
*Thread Reply:* Maybe we can discuss this after SPAAM in one of the channels. First thing would be to identify points of interest and evaluate if all together they could make a story
*Thread Reply:* Drafting any manuscript takes lot of time, it must make sense
kind of a guideline of controls to be taken into account, accompanied by nice good examples to show why each control is necessary
@Nikolay Oskolkov, how do you sort through the hits from krakenuniq to decide what to put into the MALT custom database for each project? Do you check for a list of “usual suspects” pathogens?
*Thread Reply:* We apply depth and breadth of coverage thresholds reported by KrakenUniq for eliminating obvious false-positive organisms and use all organisms reliably detected in at least one sample for building a project-specific MALT database. Ideally those thresholds should be sample-specific, we are working on it, there are some ideas but so far hard thresholds
*Thread Reply:* Do you have your workflow published or deposited somewhere for reference? We have done a similar approach but with 2 malt runs, one with a tiny database with our species of interest to filter out anything that is definitely not what we are looking for. Then extract all the aligned reads, and run them through a big Malt databse with all bacteria... but as you said, the memory usage is a big problem. Your way of flipping the approach around is much more HPC friendly
*Thread Reply:* It is going to be published soon. I think during this conference I heard a similar line of thinking a few times already, i.e. using some way of pre-selecting reliable candidates and building MALT database on them. Glad to hear that other people also thought about it
@Nikolay Oskolkov @Nico Rascovan get a room (in gather town, afterwards ;) )
*Thread Reply:* Soooooorry for that! I’ll control myself next time… 😉
Hi everyone thank you for the opportunity to share my study. Here is my paper related to my talk https://doi.org/10.1038/s41598-021-92981-8
Could you please post how to get to Gathertown? Sorry I missed it yesterday
*Thread Reply:* Your child is very cute 🙂
SPAAM3_Islandvibe https://gather.town/invite?token=VrLwFkWq SPAAMiscoolio2021
Another super day!! Thanks so much everyone! 🎉 Off to eat an eclectic meal to confuse future bio-arch researchers 😉 (assuming we don't fix these issues in our life time)
Hi all, I've attached the ethics form I discussed yesterday! Feel free to reach out with any feedback/suggestions 🙂.

Day two drawings from @Petra Korlevic 😍
https://twitter.com/petrathepostdoc/status/1433507735673376771?s=19
 }
}

*Thread Reply:* This is absolutely awesome! How do you do it? I mean is it drawn on a computer or by hand and then digitized? You are amazing!
im here finishing the comics while everyone is in gathertown so i cant confirm if there were bacteria with little silly hats
*Thread Reply:* There were people sprites with silly hats!
@Betsy Nelson has proof ;)
*Thread Reply:* close enough then! too bad you cant make your avatars in little bacteria
*Thread Reply:* That's an awesome idea... Should Pitch it to them!

@Petra Korlevic you’re so great!
How do you do to listen and drawing ?!? I barely can listen and hang my laundry at the same time
it all started as a challenge to myself a few years ago while trying to take notes in a creative way and not fall asleep on a retreat 😬
people liked the doodles so much it motivated me to continue from there
@Petra Korlevic do you teach classes because I would totally attend!
*Thread Reply:* no but i was definitely thinking about something of the sort at some point 😄
closest thing is we have some school engagement on flying insects so a good excuse to teach kids how to draw silly bugs
*Thread Reply:* I’d sign up for a silly bug class too 🐝
*Thread Reply:* You could probably set up a patreon 🤔
*Thread Reply:* hah wonder if there would be a crowd for a sci comm style patreon 😄
*Thread Reply:* I really think there would be! There is bigger bigger outreach requirements with grants etc, and so there is interesting interest in ways of doing so.
Certainly in our department I'm in there were lots of volunteers to help out drawing stuff in the archaeological science colouring book: http://christinawarinner.com/outreach/children/adventures-in-archaeological-science/

*Thread Reply:* oh i love the colouring books 😄 though i never finished my copy of it
@channel! Good day spaamers!! Last day of #SPAAM3 🦠!! Let's keep up the great talks and discussion for the last session: Tool up or die 💻🦠!! See you all in 20 minutes!!!
and the link again: https://us02web.zoom.us/j/82064835502?pwd=Q0NzVWM3cGtnRm9Vb2pyK3Vha00zdz09 ⬅️ Meeting ID: 820 6483 5502 Passcode: 831478
Paper from Irina: https://journals.asm.org/doi/full/10.1128/mSystems.00080-18
@irinavelsko super interesting and important work, thank you! When you say "detected" or "not detected", this would depend on you filtering strategy, right? E.g. Methaphan gives you normalized abundance values from 0 to 1, how do you decide if a species is present if it has e.g. a value of 0.1?
Fantastically pedagogical explanation of LCA from @Maxime Borry!
@Maxime Borry how does the alignment.bam look like? I mean the one you get out of "sam2lca analyze input.bam > alignment.bam"?
@Maxime Borry Have you noticed a better taxa detection with LCA directly computed on the sam file than with LCA done from a blast of reads presenting multihits after a mapping step (e.g. process done by Megan)?
Thanks @irinavelsko for the talk! Question open to all those using databases of complete genomes (and doing microbiomes): how do you deal with genome lengths? Any normalizations?
*Thread Reply:* Isn’t Braken preforming some type of normalization by genome size?
*Thread Reply:* If I remember well it does not, it tries to assign the reads in higher taxonomic ranks to an actual species but no genome lengths accounted
*Thread Reply:* This is really interesting! How do you account for reads that cannot be classified to the species level (and presumably then you don’t know the genome length)? Do you only focus on normalizing by those taxa that you do have good classification for?
*Thread Reply:* Yes what I do is to run Bracken and then get the reads assigned at the species level for the normalization. Reads that are not placed by Bracken (after Kraken classification) in a species remain unaccounted (unfortunately).
*Thread Reply:* We have been doing the same as @Claudio Ottoni mentioned: normalizing by genome size after Kraken. For reads that could only be assigned to a higher taxonomic level, we used median genome size for this taxonomic group (which is not ideal but in our mind still better than doing nothing). However, since starting to use Braken we stopped normalizing. Maybe @Adrian Forsythe can comment on the internal normalisation process of Braken
*Thread Reply:* Sharing input on Bracken from a former lab member (Jaelle Brealey): Bracken takes into account the kmer size used to build the kraken database and the read length, and does some fancy statistics to “derive probabilities that describe how much sequence from each genome is identical to other genomes in the database, and combine this information with the assignments for a particular sample to estimate abundance at the species level, the genus level, or above.” If they claim they estimate species abundance, not just read counts, then they should by default take into account read length, genome size, etc.
@irinavelsko I have a not-so-fully-formed question/thing I need clarification on: So some of the species on your graph (notably Bordetella pertussis) are listed as "under-detected" are species I tend to routinely see in my outputs (which I highly suspect are false positives based on soil samples and context). Does this just mean I'm using a different database? Part of the problem is that I'm not the person who set up the programs for our lab so the database stuff seems like a bit of a black box for me.
Question to Maxime: Have you tested:
- how much diskspace takes to make a bowtie DB of the NCBI nt database, and how much RAM it takes to load it for the analyses?
- How many folds bigger get the bam files of the results (when using the -a parameter), compared with the initial size of the fastq of a metagenome
- Have you benchmarked the results of using NCBI nt DB compared to KrakeUniq on the same Database?
*Thread Reply:* I could perhaps comment on the first question, to my experience it is 1TB of RAM for Bowtie2 which is much less than what is needed for MALT (3.5 TB of RAM). Let us see if Maxime can confirm it 🙂
P.S. after double-checking it was 1.3TB disk space for storing Bowtie2 index, and 3.3 TB of disk space for storing MALT database
e.g. you could actually run MALT to align it
with sam2lca in stead of MALT
@Maxime Borry have you come across this pipeline? https://github.com/frederikseersholm/getLCA also lca from a sam file

*Thread Reply:* No problem, think the publication date was around 2016. The documentation is terrible. It uses a python script and spits out a txt file of matches and read counts. As a user and not really an understander of these tools it was quite a confusing process for me 😂 I also never managed to get it to report anything that wasn’t to sp level, but again that could very much be user error!
super cool tool @Maxime Borry, potentially a game changer. Bowtie2+sam2lca should replace MALT as it should be potentially much faster and less resource demanding compared to MALT. I would be definitely interested in a potential collaboration 🙂
@Maxime Borry could you please share the link to the blog entry you mentioned? I couldn’t find it
https://maximeborry.com/post/rocksdb/

*Thread Reply:* I thought it was about sam2lca, but maybe I misunderstood
*Thread Reply:* This is part of the "guts" os sam2lca
Not sure how long I’ll be able to stay today but just wanted to say a HUGE thank you to all SPAAM3 organisers and presenters! This is such an amazing workshop and a fantastic community that sets new standards to how exchanging research ideas, tools and experience should be done.
*Thread Reply:* Thank you so much! Thanks to the spaam community for being awesome! Love y'all!:facewithcowboy_hat:
That subway map figure 🤩
A comment on Fred’s presentation. Actually AdapterRemoval was going before the QC… it was just a mistake in the slide that we missed…. 😉
*Thread Reply:* I awas about to say 😆
*Thread Reply:* Ha-ha, I do QC both before and after adapter removal, it is nice to see how QC metrics change after adapters have been removed
*Thread Reply:* We also do that in eager too... peoople weren't checking their QC reports with EAGER1, and when they started looking at MultiQC they were shocked at the before- and after- deifference
*Thread Reply:* In fact we do that too. But since the QC also included some metagenomic profilers and other tools, these tools are only run after AdapterRemoval, it is more accurate mentioning the QC after the AdapterRemoval step… 😉
@Frédéric Lemoine does the pipeline offer multiple insertion points? Could you (e.g. run eager to) generate the VCF files, and then start AMPHY from the MSA step?
@Frédéric Lemoine - Great talk! You can construct both full alignment and variant based alignment? Is there an option for partial deletion of sites to deal with the ambiguous sites we often have when working with ancient genomes where there is no call in the ancient genome but its covered in modern representatives?
Is AMPHY openly available already?
(don't need to say that one, one word asnwer is fine)
@Frédéric Lemoine why did you decide to go with raxml as your ML tree tool? @everyone are people mostly using raxml? does anyone have any experience with IQtree?
IQTree ❤️
Are we going to get to a point where unifiedgenotyper becomes unusable bc GATK has deprecated it?
Got to go, would like to express my infinite gratitude to all SPAAM organizers, it was such a warm, informative and friendly meeting, I reaaly learnt a lot, please keep doing it!
*Thread Reply:* Nikolay! It was so nice to “meet” you. Thank you for all the incredibly helpful tips and support!
DO I sense a collaborative project coming up???? VCF translator? Comparing variant callers??? 😏

Here is my script for MSAs: https://github.com/campanam/vcf2aln
Throwing the idea of a light touch mentor scheme out there……
*Thread Reply:* I think a lot depends on how the pandemic and the response to it develops
Thank you, this experience has been wonderful!
ISBA meeting Sep 21
For projects I have worked on, I/we got ethics approval from the (select all that apply)
Thanks organizers! This was a great meeting!
That was great huuuuge thanks everyone!
Quick dog walk and then will be in gather.town!
So informative! I learned so much! Thank you!
Thank you all!! So helpful and such a friendly community!!
Very nice meeting! I enjoyed it a lot. Big thanks to the organizers who did an amazing job!
Gathertown link: https://gather.town/invite?token=VrLwFkWq Password: SPAAMiscoolio2021
✨New channel alert! <#C02DCKJ54JX|no-stupid-questions> is a designated space for early stage microbe hunters to ask anything at all without judgement! Newbies can help each other and call on more advanced people to help out as needed!

*Thread Reply:* A desktop computer rolled in James’ hair?






https://en.wikipedia.org/wiki/Hockey

and final day done 🙂 https://twitter.com/petrathepostdoc/status/1433861839222448152/photo/1

*Thread Reply:* The tooth on Irina's picture looks like it has a face in it to me, and it looks like it's in bliss because it's been hugged by the calculus
*Thread Reply:* Which is perfect because of @irinavelsko's Twitter handle
*Thread Reply:* https://twitter.com/FzzyToothSweatr?s=09
*Thread Reply:* ahaha i drew one of these cozy teeth for a talk on isba and just fell in love with how silly and friendly it looked
*Thread Reply:* They are amazing 😍
*Thread Reply:* I also thought all the teeth are smiling, they’re so cute!
Thank you @Petra Korlevic!!!
heya everyone, wrapped up the event into a single poster with all speakers (and an additional spam doodle) enjoy the colorful summary!
(even at small resolution the file is quite giant)

*Thread Reply:* That's beautiful 😍
*Thread Reply:* Thank you so much for all your effort into this, it really makes these remote events to have much more impact 🤩
*Thread Reply:* glad it added a twist on the zoom meeting 😄
*Thread Reply:* Thank youuuu!!! This is wonderful!
*Thread Reply:* Would you mind @Petra Korlevic if we print this as a poster??!
*Thread Reply:* absolutely fine, i sent the link to the A0 file to to @Miriam Bravo but here is the google drive link for anyone that wants a version (file has 21MB... dont know if slack can handle that) https://drive.google.com/file/d/1Zv_i17gHQBDhwq2Weq5Q15Mheufx5_b2/view?usp=sharing
*Thread Reply:* Thank you so much @Petra Korlevic!
*Thread Reply:* You’re so talented @Petra Korlevic !
*Thread Reply:* I am speechless, it’s amazing @Petra Korlevic!
@Petra Korlevic’s amazing poster in physical form! (Apologies for my terrible photography)



*Thread Reply:* it sure adds a lot of color to the area 😄
*Thread Reply:* It really does!
*Thread Reply:* Need to get more of your doodles and stick them up everywhere 😉
*Thread Reply:* well there used to be some over in the "methods" corner since i was sitting there and doodling constantly (not sure what it is now or who sits there but it's around Janet's area)
*Thread Reply:* oooh I might go and do a hunt later and see if I can find them. They did take out a lot of stuff out htough
Hello!
For everyone who participated in SPAAM3 two weeks ago, THANK YOU SO MUCH! The talks, the discussions and the plans for future collaborations, were such a huge success and we hope you feel the same way :).
We're still editing the recorded talks and the notes from them. Once we have finalized these tasks, we will share them with attendees.
As a last request, we'd like to hear feedback about your meeting experience, so please complete this survey, it will take less than 5 minutes from your day. Your responses will impact the following editions of SPAAM.
Hi @channel ! We designed some SPAAM stickers and we will send one to you if you are interested and participated in SPAAM3. Please get in contact with me, and we can arrange the sending by post of the sticker!
