Public Channels
- # 2022-summerschool-introtometagenomics
- # 2023-summerschool-introtometagenomics
- # 2024-acad-aedna-workshop
- # 2024-summerschool-introtometagenomics
- # amdirt-dev
- # analysis-comparison-challenge
- # analysis-reproducibility
- # ancient-metagenomics-labs
- # ancient-microbial-genomics
- # ancient-microbiomes
- # ancientmetagenomedir
- # ancientmetagenomedir-c14-extension
- # authentication-standards
- # benchmark-datasets
- # classifier-committee
- # datasharing
- # de-novo-assembly
- # dir-environmental
- # dir-host-metagenome
- # dir-single-genome
- # eaa-2024-rome
- # early-career-funding-opportunities
- # espaamñol
- # events
- # general
- # it-crowd
- # jobs
- # lab-community
- # lactobacillaceae-spaam4
- # little-book-smiley-plots
- # microbial-genomics
- # minas-environmental
- # minas-metadata-standards
- # minas-microbiome
- # minas-pathogen
- # no-stupid-questions
- # papers
- # random
- # sampling
- # scr-protocol
- # seda-dna
- # spaam-across-the-pond
- # spaam-bingo
- # spaam-blog
- # spaam-ethics
- # spaam-pets
- # spaam-turkish
- # spaam-tv
- # spaam2-open
- # spaam3-open
- # spaam4-open
- # spaam5-open
- # spaam5-organizers
- # spaamfic
- # spaamghetti
- # spaamtisch
- # wetlab_protocols
Private Channels
Direct Messages
Group Direct Messages

@James Fellows Yates has joined the channel










@Meriam Guellil has joined the channel

@Gunnar Neumann has joined the channel





@Maria Lopopolo has joined the channel


@Claudio Ottoni has joined the channel
@channel what sort of (no stupid) questions would you be interested in covering if there was an ancient metageomics summer school?
Databases! How to choose, download, build
Anything else (because noone has an answer for choose 😆) ?
For sure, how quick do you need them or can I just post them here as they arrive in my brain
How to authenticate your species hits for dummies. Is a match really a match?
*Thread Reply:* authenticate what?
*Thread Reply:* Species hits, i’ll edit
I am working on trying to pull together information I learnt last week, planning for just personal use, but something more “formal” might be more helpful for a wider group
The reason why I ask is in my new position I have to set up a summer school (part of the grant its funded by)
So this will actually be a thing at some point, but need to work out content
Once I get results from EAGER and have no idea what they mean, I'll also have some "not stupid" questions 😬

Hi everyone! I’m wondering if people have preferences for variant calling tools for ancient pathogens. Never done this before!
1) If you want to use MultiVCFAnalyzer (e.g., to visualise cross-mapping) you must use UnifiedGenotyper 3.5 with ploidy set 2 2) Otherwise, AFAIK there isn't one in particular. FreeBayes is popular though
*Thread Reply:* I’ve been using freebayes. It’s very easy to use. I’ve played with the parameters to try to mimic the emit all sites option of UnifiedGenotyper (ie force it to be a genotype caller) so that I could use the vcfs with multivcfanalyzer or MUSIAL, but I haven’t been successful. I’m happy to share all of my scripts if you want.
*Thread Reply:* Yeah, you need a very highly specific format of your VCF files exactly as it comes out of GATK3.5 🙄
@Kelly Blevins you could also post on the MVA github page to put pressure on Alexander to find and properly release the VCF format agnostic version 😉
*Thread Reply:* @Kelly Blevins, that would be incredible! Thank you so much! I’ve never used a variant caller before so I’m starting from complete scratch here

Hi everybody, would be great if someone could help me. I am trying to classify test reads (generated from RefSeq viral sequences) with MALT (latest version) using as db the RefSeq viruses. Unexpectedly, I get some misclassification outside of the superkingdom Viruses, namely within Bacteria, Archaea and Eukaryota. Does someone can give me a hint, why I get a classification ‘outside’ of the database?
What exactly do you mean by 'refseq viruses'?
What has gone into your database?
Hi James, thank you for the quick reply. That is how I build the db:
wget <ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/viral.1.1.genomic.fna.gz>
wget <ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/viral.2.1.genomic.fna.gz>
wget <ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/viral.3.1.genomic.fna.gz>
wget <a href="http://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_wgs.accession2taxid.gz">ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_wgs.accession2taxid.gz</a>
malt-build -i **.fna.gz -s DNA --index viral_db --acc2taxa nucl_wgs.accession2taxid.gz
Hm ok. Have you looked at the list of non-virsus to see what they are, whether it's consistent across all samples?
I am slightly suspicious you're using nucl_wgs accession IDs, as that is just raw sequencing data and I guess could contain contamination? On the otherhand that's just the taxonomy, so you shouldn't have those sort of refeference sequences that they could align to, in your DB 🤔
- Not yet, that is the next step on my agenda…
- You are right that this accession ID’s may not be the best choice. But on the other hand since the input sequences are ‘just’ RefSeq sequences, I would have thought that this should be rather stable. Which accession ID’s would you recommend?
What version of MALT are you using?
MALT (version 0.5.3, built 4 Aug 2021)
I think that should still work
I can't remembe off the top of my head the minimum assembly build level a 'genome' needs to be to get to RefSeq ...
Maybe there is some messy WGS stuff in there? I don't know...
Thank you James, I will try it out with the indicated accession table and see if I get more convincing results… Having a messy accession table would explain the observed pattern. Let’s see. I will report it here later. James, thank you for the quick help!!!
It would be nice if someone else could chime in though, I might be talking out of my arse 😬 (@Maxime Borry?)

Hey @Samuel Neuenschwander, At what taxonomic level do you get your assignations outside of the virus clade ?
Hi @Maxime Borry, indeed from the incorrectly classified reads ~6% are outside of the db scope, e.g. not Virsues. The classified rank varies, but is often around the species level.
Interesting, I see two possible explanations: • the accessions IDs of some of these viruses match to their host (eg. human or whatever) • The RefSeq viruses database doesn’t only contain viruses
Yes, I agree. Both points are not so nice…
Does it contain phages? That could explain some cross hits depending on the database building
That is a good point, I have to check. I don’t have the data in front of me, but I also get insects… Thank you @Meriam Guellil & @Maxime Borry!
There could be a lot of insect viruses in the DB, because they’re commonly used in molecular biology as vectors for eukaryotic cells, the so called baculoviruses
Maybe contigs are taxonomic assigned in some cases in the assembly pipeline used by some researchers, and so those contigs pop up, even if the vast majortiy of contigs are from teh virus itself?
(so point 1 of what Maxime said above)
Yes, that makes absolutely sense, if an ‘entire’ db with all taxa would be used. With a virus only db however this should not be possible, if not one or the other input is messy.
I have replaced the accession file (with the one pointed at by James) and then also used other viral sequences to create the MALT db, but more or less with the same result. I am trying to nail down the problem, but first I will go on vacation 😉 Thank you fore the great help @James Fellows Yates@Meriam Guellil@Maxime Borry!
Hi everyone! There’s a discussion going on in my lab about whether or not to sample from a tooth with a pretty large, visible cavity. I’m wondering if people have any thoughts!
*Thread Reply:* Can you share a photo or sketch of what you mean?

*Thread Reply:* We have an ongoing project on bear oral health (@Adrian Forsythe can chip in) and see that sampling from within and closely around cavities produces a very distinct microbial signature, whereas samples from healthy teeth in the mouth with cavities look almost “normal” (e.g. they hardly differ from samples from healthy individuals).

Opinion question for the pathogen peeps: I'm screening samples for pathogens, currently particularly focusing on TB and Leprosy. If you see in MALT 100% identity reads mapping to either M. leprae or M.tuberculosis, and those same reads also map to for example M. avium with one mismatch (~97%), would you trust those reads as M. leprae/M. tuberculosis? 🙃
*Thread Reply:* Calling: @aidanva @Åshild (Ash) @Meriam Guellil @Marcel Keller @Maria Spyrou (off the top of my head)
*Thread Reply:* @Betsy Nelson for TB as well


I would say it depends on different factors. How many reads do you have? Are those reads distributed evenly on your reference? Are those reads coming from a low diversity region?
So I guess my point being, it is very hard to do an assessment on a single read
one need to look at different lines to see if what you are seeing is actually the species of interest.
but maybe others can chip in since I have not worked on mycobacterium identification so far

i would map your reads to the references of the various hits, compare the number of hits normalized over the reference length (even in Mycobacterium, genome lengths can vary by quite a bit) and also investigate edit distances to try to get a handle on where the reads ''''truly'''' are coming from. as with most things we do, nothing will be "definitive" but the above should give you a better idea of the affinity of the reads to each of the reference genomes, the coverage across the difference reference genomes and the edit distance plots which look the best (ie steadily declining rather than increasing edit distances to your reference)
Others might suggest doing a competitive mapping within the mycobacterium diversity, but i have not done this myself.

Thanks @aidanva and @Ian Light for your answers! Yes, I've also been looking at the evenness of coverage and edit distance (although I still need to create plots). I think my main problem (and probably everyone else's looking into Mycobacterium species), is trying to see through all the background in the hope there are some very low levels of a true signal... I've been using BWA mapping and MALT so far, and now working on extracting all the reads mapping to the MTBC clade in the MALT results as a way to filter out background noise... Any thoughts on this approach??
I also want to try stricter BWA mapping parameters. I've been using an edit distance (-n) of 0.03 for the screening, but that still picks up very deep clusters/conserved regions with lots of multiallelic sites. But I wonder if there might still be a more random distribution buried underneath these clusters. 🤔
Yeah, Mycobacteria are a harsh one. I think in this regard @Betsy Nelson @Åshild (Ash) @Kelly Blevins can share their experience working with TB
It’s great to have this channel, and I’m happy to contribute where I’m able to. But can we establish as etiquette that questions are only answered with the thread function? This way it’s easier to keep track on what’s going on and one doesn’t get flooded with notifications.
*Thread Reply:* If you are getting bothered a lot, I would highly recommend setting your notifications to mention only:
(right click on the channel name > change notifications)

*Thread Reply:* Then you will be notified when someone wants your specific feedback, but otherwise you can just look in there when you're interested
*Thread Reply:* not bothered a lot, but I would still say for a channel meant for questions and answers (rather than general discussions or announcements) it would help
Most species have their own threshold of how much sequence coverage you need to be able to identify them for sure which depends on a lot of factors and this can sadly vary a lot. If you don't have enough coverage there are a couple of other rabbit holes you can go down (specific regions, competitive mappings, masking of conserved regions, etc) but sometimes you will need to increase the coverage to be sure. Without seeing the data its hard to tell, but I am sure the MTB people have some tips up their sleeves :)
(sorry @Marcel Keller ship has sailed for this question lets do better on the next one 😉)
@Meriam van Os I echo everyone else’s advice - breadth of coverage and edit distance are really important. If you’re picking positives from MALT for downstream investigation though, I have found that the frequency of MTBC summed reads/mycobacteria summed reads (at least .25) predicts successful capture enrichment pretty well. If you’re seeing something that looks like the attached, I would say don’t get your hopes up. How deep are these libraries sequenced? And how old? Most of my experience is from screening ~500 year old remains from central Mexico for MTBC, but I’ve worked with some more recent (200-300 years old) samples from Belgium and Spain that followed the same pattern (intermittent qPCR assay positivity (IS1081, IS610); pileup at mycobacteria node in MALT after shotgun; less than 5% of the MTBC ref genome covered after MTBC capture, where the 5% is pileup at conserved regions).

*Thread Reply:* For example of a weak positive, attached is a screenshot after a MALT screening of a shotgun library. It doesn’t look great, right? A solid chunk of reads could be assigned to the MTBC node, but the majority could not be resolved further. I was able to recover a partial genome (~70% of the genome covered at 1x) from this library. So not enough for analyses, but enough to confirm MTBC and justify making another extract or library.

*Thread Reply:* What database are you using for MALT? I made (a small) one for myobacteria and friends that I’m happy to share.
*Thread Reply:* Hey Kelly, this is super helpful, thank you! We've built a MALT database with ~600 Mycobacterium genomes (MTBC and non-MTBC species). Do you think this should be sufficient? And, so you're saying that in MALT, if about 25% of Mycobacterium reads map to the MTBC, that's a pretty good indicator? I've done capture as well, samples have about 12 -20 million collapsed reads. All of them are showing over 700 MTBC reads with a 100% identity threshold. These samples were selected for their lesions and have tested positive for at least IS1081. Looking at the distribution today, so finger crossed something real is in there!
*Thread Reply:* Yes of course no problem! Yeah, I think that’s plenty. My myco database only has around 500 genomes.
*Thread Reply:* Yep, that’s been a good indicator for me. If at least 25% of the myco summed reads can be assigned to the MTBC, then I have been able to get at least a partial genome from the library after capture. So I would say it’s not a way to authenticate MTBC positivity in a sample but a way to predict a positive capture, if that makes sense. I don’t think we’ve a good enough grasp of myco diversity to authenticate MTBC with just a few reads, even if they’re evenly distributed.
*Thread Reply:* Depending on your capture efficiency, you should be able to get away with sequencing at half that depth. I’ve found that I can sequence at a depth of ~2-3 million reads after Daicel Arbor Biosciences myBaits capture (we send them MTBC genomic DNA and they make the baits and send us the kit https://arborbiosci.com/genomics/targeted-sequencing/mybaits/mybaits-expert/mybaits-expert-wge-whole-genome-enrichment/) and expect to sequence the captured library to saturation.

*Thread Reply:* Good luck! You’re working in the Pacific, right? I REALLY hope you find something 🙏:skintone2:

@Adrian Forsythe has joined the channel
Hello, I have a question. If you want to re-sequence a certain library to reach the desired genome coverage (e.g. 10X), where the targeted genome could be human, microbial, etc., which calculation do you use to estimate how many additional reads/Gbp you will need?
Of course the simple formula would be something like this: Reads to generate = [ GENOME SIZE ** (proportion of unique mapping reads from total raw reads) ** Expected read depth ] / (average read size of uniquely mapping reads)
A second option is to Run preseq, and propagate the curve it estimates. This solution is certainly better than the previous one, as the % of duplicate reads is not linear.
Do you have some other method that outperforms preseq for this? If using preseq, do you use any particular tuning ? Do you have a script to share? (other than the outputs we can get from nf-core/eager)
*Thread Reply:* @Felix Key has a nice little script that parses PreSeq output to give you more precise information: https://github.com/keyfm/shh
*Thread Reply:* However looking at the README it might customised for eager1 😕
*Thread Reply:* So you might have to do some reconstruction
*Thread Reply:* Looks like this:

*Thread Reply:* (CF == cluster factor == duplication ratio)
*Thread Reply:* Still PreSeq though
*Thread Reply:* It looks liek a lot ofnf-core pipelines still use PreSeq actually, so I dunno if there is abetter mehtod
*Thread Reply:* ok, great. Thanks a lot James. I think that building a plot like this will be enough for a first try. We were just discussing today with @Pierre Luisi if in addition to the expected distinct reads curve we could also estimate if the genomic positions covered are evenly distributed along the genome or if there is also some sort of asymptotic curve of the chromosomes/regions/breadth of coverage while increasing the sequencing depth, that does not fully follow the same trend than the preseq curve…
*Thread Reply:* you can also have a look at nonpareil https://github.com/lmrodriguezr/nonpareil
*Thread Reply:* Two caveats about nonpareil:
1) It's more for metagenomics rather than genome coverage for a target genome 2) Only supports uncompressed FASTQ files, which sucks baaaaad
*Thread Reply:* I see. Incredible that there are still tools using uncompressed fastq files out there… I got the impression it was more metagenomics oriented
*Thread Reply:* but it’s good to know it exists
*Thread Reply:* A LOT of metagenomic tools still only accept/produce uncompressed files
*Thread Reply:* It was crazy, Maxime and I went through loads recently. But I think with modern data, you don't need to sequence much...
*Thread Reply:* still, disk space is way too expensive to spare it in something like this
*Thread Reply:* Either the devs have 💰
*Thread Reply:* or just tiny assemblies

Hello everyone :)
I am wondering what the SPAAM community thinks of the different releases of Kraken2. I did my master's project on dental calculus and the first time I run Kraken2 (v. 2.0.8 beta) / Bracken I got almost 10000 taxa identified in the dataset. I recently analyzed (almost) the same dataset again, using Kraken2 v.2.1.1 and only got about 2500 taxa identified. From what I understand, this most recent release of the software works a bit like KrakenUniq in reducing the false positive rate. Do you think this change could explain such a big difference in the number of taxa identified? In your experience, do the most recent versions of Kraken2 indeed perform better? Would there be any acceptable reason to keep using an old version?
*Thread Reply:* To add to that: With the new version of Kraken2, we recover considerably fewer oral taxa from our community (40 vs 120).
*Thread Reply:* Hi everyone! I've been thinking about this question a bit since I'm currently using Kraken2 but have seen how Kraken1+KrakenUniq was a preferred alternative during SPAAM3. I found the github page with the changes in each version released (https://github.com/DerrickWood/kraken2/blob/master/CHANGELOG.md) but I don't understand it well enough so I don't know if these changes are more likely to affect false positive detection. Would it be possible to get advice on this? A friend and I in our group are the only ones doing metagenomics and we're kind of towards the end of our PhD so now is a good time to decide whether it's ok to stay with Kraken2 or if we should just reconfigure our reference database for Kraken1 and KrakenUniq. If Kraken2 now behaves a bit less wildly with false positives that would be a faster solution for us since all that's needed is a software update!
*Thread Reply:* @James Fellows Yates if you get a second could you please do your tagging people who might know on this post? Obvs no rush!
*Thread Reply:* Oooff...
*Thread Reply:* I'm not really sure. Generally I would use the latests tools, but I've really used kraken2...
*Thread Reply:* @Nikolay Oskolkov or @Maxime Borry maybe?
*Thread Reply:* @Nikolay Oskolkov was doing some comparisons between KrakenUniq and Kraken2 if I remember correctly.
*Thread Reply:* But the official word is that Kraken2 (since version 2.1.0) with the --report-minimizer-data flag should produce the same results as KrakenUniq
*Thread Reply:* Thanks Maxime 🙂
*Thread Reply:* Thanks everyone. Any first hand experience comparing the new version of Kraken2 to the older one? I am happy about the reduction of the overall number of taxa detected (as obviously the previous version was recovering lots of spurious hits) but the loss of 2/3 of oral taxa is scary…
*Thread Reply:* Did you use the same database & database version?
*Thread Reply:* Maybe just many more genomes added and pushed hits further the tree?
*Thread Reply:* No, new database, so we are not sure what is causing the effect. Have not tested systematically, as simply not enough time for that
*Thread Reply:* Am hoping someone has the answer 🤪

*Thread Reply:* Hi guys, a lot of things to say here, sorry for being late to the discussion. First, I do not think that newer versions of Kraken2 are more accurate than the older versions of Kraken2 @Katerina Guschanski. Instead, the growth of databases (as @James Fellows Yates asked about) with time usually results in fewer detected taxa (at the same depth of coverage threshold). Second, newer versions of Kraken2 themselves do not reduce false-positive rate @Markella Moraitou if you do not filter your output with respect to the breadth of coverage, which is provided ONLY if you use the --report-minimizer-data flag as @Maxime Borry mentioned, please note it is not a default flag. Third, even with --report-minimizer-data the breadth of coverage delivered by Kraken2 is very different from the one from KrakenUniq (if you just look at the number of unique kmers stats), however, they are correlated. In my case, after I checked ~10 human + non-human samples, the correlation slope was ~0.7 meaning that if one used 1000 unique kmers as a threshold for KrakenUniq, this should corespond to 700 unique kmers threshold for Kraken2 in order to get more or less comparable final lists of detected taxa. Fourth, if you carefully examine Kraken1 / KrakenUniq and Kraken2 papers, they clearly write (in the Kraken2 paper) that the database search they have implemented in Kraken2 (approximate minimizer search instead of exact kmer search in Kraken1 / KrakenUniq) reduces specificity of classification (how much - not clear) but offers instead superior speed and memory advantages. Moreover, the authors also mention the effect of "collision" of minimizers corresponding to different kmers (a minimizer is just a substring of a kmer), which introduces some "randomness" in the taxonomic classification from one database to another. In other words, no two Kraken2 databases are identical (obviously not good for reproducibility), so even if you use the same ref genomes, two databases built at two different time points might result in different classification results. In summary, after extensive testing Kraken2 vs. KrakenUniq I am still not convinced that the speed and memory advantages of Kraken2 can compensate for the reduction in specificity
*Thread Reply:* Brah... That would be a perfect blog post, literally just that... @Shreya @Ele 👀
*Thread Reply:* Wow, so cool and helpful @Nikolay Oskolkov! Thank you so much for the explainer!
*Thread Reply:* Thanks @Nikolay Oskolkov (and everyone else who responded)! I was not aware that the increase of database size could have this effect!
*Thread Reply:* @Maria Zicos think this will be of interest to you. @Nikolay Oskolkov as always you explain things so nicely, thank you! And yes if you fancy a writing a blog post we would love to host your expertise! Let me know and I can send you some details 🙂
*Thread Reply:* @Nikolay Oskolkov comment prompted me to have a look at the Kraken2 paper again: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1891-0 One additional point that I should point in favour of Kraken2: it uses spaced-seeds, meaning that it allows for mistmatches (7 out of 31 by default) in the minimizer, which AFAIK, is not the case for KrakenUniq. In KrakenUniq, no exact kmer match = no detection.
*Thread Reply:* That's really interesting, @Maxime Borry. So this could be one of the reasons for higher number of taxa detected by Kraken2
*Thread Reply:* Could be one of the reason, yes. You can also adjust this parameter to allow the number of mismatches that you want (from 0 to 0.25**minimizer length).
*Thread Reply:* @Maxime Borry for KrakenUniq / Kraken1 I would say "no exact kmer match = no assignment of the kmer", which does not necessarily mean that a read is not assigned since a read can have a number of kmers and they all "vote" for a particular taxon. But generally agree that Kraken2 is more permissive and KrakenUniq / Kraken1 is more conservative.
Hi, all! What is everyone doing to decontaminate skeletal elements before powdering? Does everyone still UV bone samples for 15-30 minutes per side? Is there any concern that doing so does more harm than good?
*Thread Reply:* Uving isn't actually AS bad as some people thingk.
*Thread Reply:* @Christina Warinner often refers to this paper: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0013042
*Thread Reply:* It can be bad if you have wet samples + UV is strong and right next to the sample (IIRC)
*Thread Reply:* We tested UV only vs. EDTA wash only on a small subset of samples and ended up doing both, as both were removing some parts of contaminants but having little effect on endogenous (oral, using DC samples). Not very systematic results of this not very systematic experiment are presented here: https://academic.oup.com/mbe/article/37/10/3003/5848415?login=true
*Thread Reply:* We also forgot to put away our tooth samples from the lab bench once and they ended up under ceiling UV for 2 h, without any advert effects on the host endogenous content. We never published this faux pas (surprise...)
*Thread Reply:* sounds like a good anonymous twitter post 😉
*Thread Reply:* #HonestMethods
*Thread Reply:* Thank you both so much! This is very helpful 🙌:skintone2:

*Thread Reply:* With UV, it matters a LOT how close your sample is to the UV bulb. The strength really drops quickly off with distance. The main advantage of using UV in a room is that it kills all the microbes on the surfaces and so keeps the room low biomass. However, overhead UV does very little to short aDNA fragments, especially if the sample is dry and not close to the UV bulb. I think it can probably mostly be skipped. I think the EDTA wash is more useful
I have a question regarding reference sequences: doing my first steps in microbiome analysis, I naïvely thought that you could go on https://www.ncbi.nlm.nih.gov/genome/, search for your species you are interested, and download the fasta under “Reference genome”. It turns out this was wrong, e.g., for Streptococcus sanguinis, where the reference genome is SK36 but ncbi directs to SK405, which is not even a full genome. So if downloading bigger numbers of different reference sequences, where do people find them without going through the literature?
*Thread Reply:* Mmmm, I would have done the same thing as you did…. You can also check all the assemblies of a species and their sizes in bp and chose one in the median size o something like that…
*Thread Reply:* thanks, in the meantime I also noticed that even for Yersinia pestis the “reference genome” is not CO92 but A1122 (assembly), so I’m wondering if this should be used at all
*Thread Reply:* That’s strange… It was CO92 not long ago… Nevertheless, CO92 was a very bad reference to use anyway… haha

Hi all! I’m sequencing aDNA libraries prepped following a protocol derived from Rohland et al 2015's partial UDG protocol on an Illumina NovaSeq platform. The samples aren’t pooled; each library has its own Illumina adapter. I’m finding that all of my R2 reads contain 20-60% single-G repeat sequences (basically 40 Gs in a row repeated dozens to thousands of times). Has anyone seen this before, and have any idea on how to remedy it for future sequencing attempts? Thanks in advanced!
*Thread Reply:* Yes, poly-G tails are common on Illumina 2-colour chemistry machines (NextSeq/NovaSeq)
*Thread Reply:* https://github.com/OpenGene/fastp
More specifically: https://github.com/OpenGene/fastp#polyg-tail-trimming
*Thread Reply:* fastP has a specific command to clip them off (we use that in nf-core/eager)
*Thread Reply:* More info: https://sequencing.qcfail.com/articles/illumina-2-colour-chemistry-can-overcall-high-confidence-g-bases/
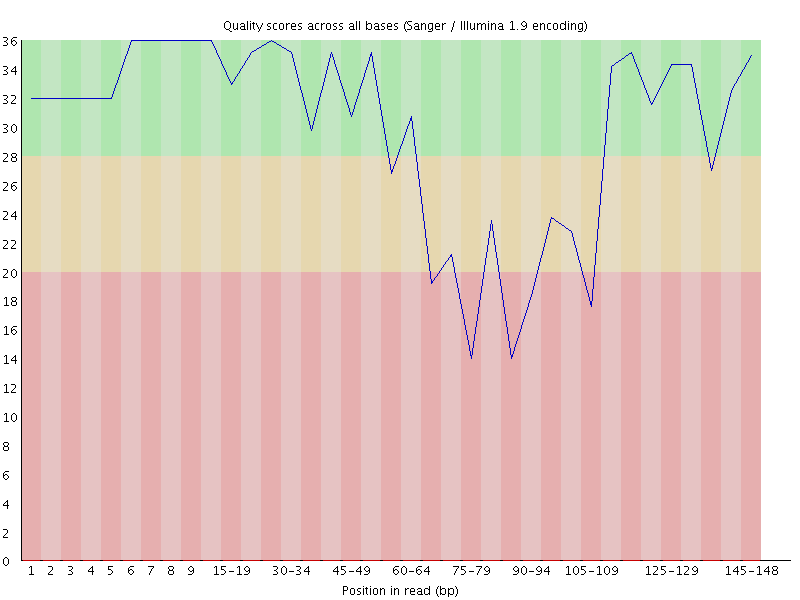
*Thread Reply:* Thank you so much for the info, @James Fellows Yates! Good to know it’s not just my libraries.
*Thread Reply:* Nope, perfectly normal 🙂
*Thread Reply:* Wondering why this would affect predominantly R2 reads. Any idea?
*Thread Reply:* End of the chemistry reagents, more out of sync cluster, meaning the camera can't work out what colour it is, resulting in 'nothing' to be detected
Hello all, I am an archaeologist doing research with aDNA for my dissertation. This is my first experience with this type of research and I am focusing on microbiome analysis from historic calculus samples. I have just received my sequenced data from the lab and I wanted to ask if either 1) someone might be willing to help me get started with analysis, and/or 2) might know of anyone/anywhere that might offer analytical services for this type of research (ideally someone who would work with me collaboratively so I could learn as well)? I am working with a microbiologist at my university, but there is no one on campus with aDNA experience, and my efforts to find external help have not been successful. I've discussed some particulars of my project with some other graduate students I met at SPAAM, but many of the programs are already setup for analysis on their lab network. Using my research questions as a guide, I have loaded all of the necessary programs for analysis to my computer, I'm just not sure where to go from there in getting things setup. I am a beginner at this, but I am eager to learn and I appreciate any guidance that anyone is willing to provide. Thank you!
*Thread Reply:* You could check the summer-school we made this year to get you started: https://github.com/mpi-sHH-SummerSchool/dag-material
*Thread Reply:* and https://youtube.com/channel/UC4ieuUEHUqYQGQF_DKIwcKA

*Thread Reply:* Thank you, I will definitely check out those resources.
I feel like there are lots of Krakenuniq questions, do we need a whole channel just for it? I’m interested in building as comprehensive a database as possible. The krakenuniq default “nt” option is only the microbes in nt— should I be manually downloading the entire “nt” from the blast server, running dustmasker on it, and kraken-building it from there?
*Thread Reply:* Nah 9 domt think it's necessary, would be overkill. What would be good is a blog post or community contribution to the krakenuniq docs
*Thread Reply:* @Shreya and @James Fellows Yates, yes, this is exactly what I do. I download the whole blast nt database and build a KrakenUniq database out of it. This however would only be beneficial (compared to the microbial NT from KrakenUniq) if you want to also screen for eukaryotes in addition to microbes. However, with sediment aDNA becoming very common (next will be catching aDNA from the air of Denosova cave via filters 🙂 ), adding eukaryotes to a database seems unavoidable to me. The problem here however is the very poor quality (or unequal quality) of eukaryotic (more specifically, mammalian) reference genomes in the blast nt database, this might lead to such mammals as horse, wolf, mouse and pig always present if you classify or align reads from another mammal (which is a pure artifact of unequal quality of mammalian ref genomes in the blast nt database). However, the poor quality of mammalian ref genomes in the blast nt database makes it technically possible to run classification or alignment (since the whole blast nt is only ~300 GB so not so much RAM is needed). If one needs to use good quality ref genomes from ~300 mammals, that would be close to impossible to use for alignment or classification (in terms of memory resources, because it would require a few TB of RAM)
*Thread Reply:* Well, I am back with more questions to annoy you with, apologies and thank you in advance @Nikolay Oskolkov! I do indeed want to include eukaryotes--humans in particular and at least being able to detect a potential mammal! I have downloaded nt.fna.gz and run dustmasker on it and now realizing I need to make the seqid to taxid mapping file in order to build the database. If I download nuclgb.accession2taxid and nuclwgs.accession2taxid, concatenate them, and pull the accession and taxid columns, would that work? Seems like the krakenuniq-download command would handle all of this for me but leave out the eukaryotes…
*Thread Reply:* Oh dear and I have to add a third column if i want to use the --taxids-for-sequences and --taxids-for-genomes! 🤯
*Thread Reply:* @Shreya yes, you need nuclgb.accession2taxid and nuclwgs.accession2taxid files but also names.dmp and nodes.dmp files, if I remember correctly. You know what, the simplest would be if you use
kraken2-build --download-library nt --db FULL_NT
this will download the full NT (including eukaryotes, invertebrates etc.) with all the mapping files needed. Despite you use Kraken2 for this purpose, you will build the KrakenUniq database using something like this:
krakenuniq-build --db FULL_NT --kmer-len 31 --threads 80 --taxids-for-genomes --taxids-for-sequences
@Abby Gancz you might also find this thread useful because you are also building a KrakenUniq database
*Thread Reply:* 😮 I hadn’t even thought of using kraken2 to download!! Brilliant!! Thank you so much Nikolay!!
Hi Friends! I have a question about library method choice for coprolite metagenomics, which I feel is applicable to general experimental design. My background/setup: I am doing dietary/paleoenvironmental metagenomics on ancient sloth coprolites for my PhD. I started the work with 7 samples, experimenting with extraction methods, and doing double-stranded libraries, which I screened. I determined the best extraction method from those (PowerSoil kit > Plant mini kit or Dabney adapted for tissue, which is the standard my lab uses for non-bone). My conundrum: I have now got five more coprolites, which I extracted only with the PowerSoil kit (because there's not too much point with the other methods). My group is looking into changing from Meyer-Kircher libs, potentially towards single-stranded protocols, likely SCR. I'm wondering whether I should still do these five libraries with Meyer-Kircher to keep consistency with the rest of the study, or whether it's better to just go with whatever is likely to yield more data. I feel the latter is a more responsible a way to use up a paleontological sample (always maximise outputs), but I'm worried that maybe using a different library protocol, especially moving from double-stranded to single-stranded, will impact the DNA community in the sample quite a bit and make these new samples not comparable with the previous ones... My questions to you all: Are some of you working with single-stranded libraries on metagenomics? How has that impacted your results? Does recovering ssDNA do cool things for detecting more taxa? Have you had trouble with mixed datasets of double and single-stranded libraries? What would you advise me to do? Do you think my choice could impact "publishability" of the work? In an ideal world, I think my instinct would be to try these new libs with the ss protocol, and if it improves yields etc. as expected, re-do the previous 7 samples with this new protocol too, and re-screen them. Only this chapter is currently not a spending priority (I'm doing a lot of popgen stuff too) so I don't know if I can afford re-building and re-sequencing the old 7 samples.
Hi @Maria Zicos
It's a very good question. No one has done a systematic study for ancient Metagenomics (low hanging fruit for a good publication I think ;))
For calculus I did a tiny bit of experimentation and it didn't make much difference when doing microbial genome construction (both very old and more recent).
It depends on your question ultimately I think. Do you want to do Metagenomic de novo assembly? In this case having DS libraries are probably better as you get more longer (still ancient!) reads, whereas SS will increase the proportion of very short but unusable reads for assembly.
However if you're looking for dietary DNA or host DNA, it would make more sense maybe to Indeed switch to SS as you will pick up a greater proportion of true endogenous DNA, which you can use for reference based mapping approaches.
I don't know of any study that estimates the amount of increase you may get for SS. I would maybe refer you to my normal poop question person @Alex Hübner, but he often is dealing with samples with unusual preservation. I don't know if he has SS libraries

Hi @Maria Zicos, That’s indeed a very interesting question and I guess no one has answered yet. I agree with James’ classification of the use of DS and SS libraries. If you are mainly interested in doing reference-based analysis, you should try SS libraries, however, they won’t be off much help for assemblies. Many of the palaeofaeces samples, we have processed in our lab that relatively good preservation but also were excavated at archaeological sites that favour preservation (low-water exposure environments) and we always only did DS libraries. Regarding the design of your experiment, in case you figure out that SS libraries are far superior over DS libraries, you should go back to re-process the old coprolites, too, if possible. You have a relatively low number of samples in your study and consistency in the preparation would increase the power for statistical analyses.
Is there any reason you could not perform Kircher et al. 2012 style dual-indexing with indexing primers of different lengths? For example, could I use a forward indexing primer with an 8 nucleotide index and a reverse indexing primer with a 7 nucleotide index?
*Thread Reply:* I don't think there would be an issue with building it (although I also might be wrong there, idk), but I think it might make data processing more difficult? I could be wrong, but I would imagine you would need to only use the first 7 nucleotides of the 8-nt index when demultiplexing (which could be an issue if that last base is the only difference between two indices) and then you'd have to trim an additional base
*Thread Reply:* This is what I am not sure about, because I have no experience with demultiplexing. I thought maybe as long as you specify the index seqs in the sample sheet it wouldn’t matter if they were different lengths.
Does anyone know how the spreadsheets that come out of the HOPS pipeline are created? We just noticed that the species listed in these spreadsheets aren't always the same when comparing different runs with different individuals, and read numbers and species listed in these spreadsheets are also not always the same as the read numbers and species present in the rma6 file for the same library (which I'm guessing has to do with that not being filtered through yet?). At first I thought it was maybe just whatever species had hits were listed in the spreadsheet, but a lot of the time there are 0 reads listed for some of these species, so I'm not sure. Is it that there are some species hard coded into the spreadsheet and others that just make it into the spreadsheet when there are hits in the rma6 file? Any insight on how the program decides which species and reads make it into these output spreadsheets is appreciated, thanks!
Answer from @aidanva and I (she's sitting next to me)
1) rma6 contains everything - consider this raw 2) hops will only report hits of stuff your taxon list that you specify. 3) However it also looks below that node (I think two levels) and report those as well (depends on the discuss). With the MALT LCA it may not push reads higher up the tree if it's unique at that node 4) Megan may not agree with hops because hops does additional filtering by default such as destacking and deduplication that will reduce the numbers of reads
Also Aida says in a given HOPS table, it will show all taxa that has been found in the run across all samples (even if that particular sample doesn't have his to it)
Someone remembers the explanation of why “porcine type-c oncovirus” is a typical taxonomic hit in metagenomics?
*Thread Reply:* If it’s a guessing game, I would place my bets on the endogenous retrovirus explanation (apparently it’s a virus that can be integrated in mammalian genomes), and/or a genome contamination (since Sus scrofa appears quite a few times in Conterminator)

*Thread Reply:* My response too! I also wonder if it could be a model organism or something...
*Thread Reply:* But more likely the retro virus
*Thread Reply:* it is just that I have a group of saliva samples, all taken and processed in the same way, but I only get the hit in some of them… so that’s the tricky part… Once would expect to find them in all or most of them… But well, of course there are many technical biases that could explain this as well…

Hello everyoneI'm writing to you because I'm doing my master thesis on a permafrost analysis. I am working on samples of a very old permafrost soil from Greenland. I have extracted some DNA from it. However, I had a very small biomass and I don't think I have much DNA. After extracting the DNA I did PCRs on the 16s of the bacteria to check if there was any in my sample and I had bacterial DNA in my samples. After that, I wanted to know the DNA concentration of my extractions.
And I could get these results:
> Sample Concentration (ng/µL) A260 260/280 260/230 > 226 4.8227 0.096 2.119 0.088 > 227 6.063 0.1213 2.073 0.044
All the numbers correspond to what we expect (very low absorbance at 260nm, but i think it is normal the biomass is very low and even more the DNA) But the 260/230 ratio is very strange, it should be around 2, and here it is really tiny. Have you ever had this kind of case with ancient DNA? What is also possible is the presence of many soil phenols in my sample. But could the presence of degraded ancient DNA have any influence on this ratio?
Thank you in advance for your answers!
*Thread Reply:* **My project consists in sequencing the metagenome of the organisms still living in this soil.
*Thread Reply:* Hi @Louis Lhote that's somewhat to say. A first caveat is that the vast majority of people here so shotgun sequencing not (16S) PCR
*Thread Reply:* For the ratios I know @Zandra Fagernäs @ivelsko might have some knowledge as they've talked about it in our group meetings in the past.
More specifically for sediment in general: @Pete Heintzman @Vilma Pérez @Barbara @Anan Ibrahim @Linda Armbrecht might have some experience

*Thread Reply:* Hi Louis, the low 260/230 suggests one of 2 kinds of contamination: guanidium salts, or protein
*Thread Reply:* Both can be cleaned up some by washing, but with such low DNA concentrations you risk losing most of it
*Thread Reply:* Hey, I plan on doing some shotgun sequencing afterwards. But the PCR was only to verify the presence of live bacterial DNA (the pcr gave fragments of about 500 bp which excludes ancient degraded dna) I want to be sure of the quality of my DNA before sequencing it Thanks!
*Thread Reply:* That's really low for post-PCR values though. How did you clean up the PCR product before you took the 260/280/230 readings? And what did you take the readings with? NanoDrop? Qubit?
*Thread Reply:* Ok thanks! @ivelsko do you think it is still possible to do a sequencing with this kind of contamination ? This is only the raw result after extraction of the dna from the soil. The PCR was only used to ensure the presence of large dna in my sample. I calculated this value with a nanodrop
*Thread Reply:* Ah, ok. With values this low, the NanoDrop isn't very accurate and it's better to take readings with a Qubit. Do you have access to one? If so, re-run your extracts through that and you'll have much more accurate values
*Thread Reply:* If you can't use a Qubit, I think you can still proceed with library building with this little DNA
*Thread Reply:* The subsequent clean-up steps and amplification should be enough to remove the contamination and bring up the DNA concentration
*Thread Reply:* @ivelsko Thank you for this information! I normally have access to a Qbit. I will try to see what I can get as a value with it. What is the risk of doing a sequencing without cleaning my sample? Can it create bias?
*Thread Reply:* The main issue will probably be poor library construction, so your library might be biased and then your sequencing results will be too. It sort of depends on the method you use for library construction, some will likely be more affected than others
*Thread Reply:* Since you're looking at the aDNA you'll probably be using the Meyer&Kircher protocol? I think that will be less affected than say the Nextera kits that use an enzyme to shear the DNA
*Thread Reply:* @ivelsko I'm actually going to do a Pacbio metagenome sequencing (I've never done one so I'll check with my lab what they use). The goal is to have the dna of organisms still living in the soil.
*Thread Reply:* I've never done any PacBio sequencing, so I'm not familiar with what they want for input quality. It's definitely good to ask the sequencing center what they expect of sample quality for high-quality sequencing data
*Thread Reply:* @ivelsko ok thanks! I'll ask

*Thread Reply:* Hi @Louis Lhote What type of material is your sediment? i had a similar problem with sediment samples from the deep layers (~20m). They had a particularly high level of clay content which i presume always affected the ratio readings of my nanodrop.
*Thread Reply:* Hi @Anan Ibrahim it is permafrost core so a very dry and cold soil. Thanks for your response! I think it is the same problem

Hi! in the lab we advanced quite a lot in processing our aDNA data with nf-core/eager. It is going quite well, but we see some bias and some metrics (with fastQC after clipping and damaeprfiler) that disturb us. We are now in the great moment when you need to undestand the effect of parameters at the different steps. I have a couple of specific question about parameters for Adapter removal and damaprofiler. Better to ask here or in nf-core/eager slack channel? Thanks
*Thread Reply:* If it's specifically questions about Adapter removal and damage profile, this is not eager specific so here should be fine
*Thread Reply:* Great! so, as a starter...
- in nf-core/eager the minimum adapter overlap required for clipping is set to 1 by default, when usually it is preferred the Adapter Removal default value (0). Why did you choose such a default parameter value??
- Also the Specify minimum base quality required for clipping is by default 20. Some people prefer to lower down that threshold down to even 2, so they keep potentially informative reads here and non informative will be removed when mapping because of the mapping quality. What are you thoughts on that? Thanks 🙂
*Thread Reply:* Uhh, can you clarify number 2 - do you mean quality clipping?
*Thread Reply:* You say adapter overlap twice
*Thread Reply:* But 1) is a left over from eager 1. There was in house testing that suggested 1 worked best. I personally don't think it's a very good idea but no one has complained so far. It's more aggressive but cleans up reads more, I guess.
*Thread Reply:* For quality trimming 20 refers to base quality score. I've never heard people going down to 2, that doesn't make sense to me because that means you would keep extremely low confidence base calls in your reads. I've only heard people go higher than 20...
*Thread Reply:* You were right for 2... base quality 😬
*Thread Reply:* for 2). I totally agree with you, and I will stick to this. but here is a quote from a Science paper (as usually digging into the Supplementary Material to get those details)... "AdapterRemoval v1.5.3 (60) was used to trim Illumina adapter sequences, leading Ns (-- trimns) and trailing quality 2 runs (--trimqualities --minquality 2) from both single- and paired-end reads."
*Thread Reply:* Thanks James, so I will definitely increase the min adapter overlap and I'll stick to the min base quality of 20🙌
*Thread Reply:* Which paper is that? That looked like it could be a typo
*Thread Reply:* Quality 2 runs sounds weird too
*Thread Reply:* But yeah no harm in increasing the number of overlap at all.
*Thread Reply:* Do you want some names? haha doi:10.1126/science.aav2621
Yes I was wondering too if it could be a typo. But Nico then told me it could be to follow the strategy I mentioned before of not be so stringent at that step and remove later when mapping
*Thread Reply:* yes, as far as I remember some people in CPH where using --minquality 2 for AdapterRemoval, I guess maximize number of reads that get mapped, despite the quality score… But I agree on keeping quality filter at 20 for our analyses
*Thread Reply:* I guess it depends how much coverage you have.
If you have low coverage it massively risks getting false SNP calls. If you have higher coverage is not as bad, maybe it's worth the trade off to get ultimately higher confidence
*Thread Reply:* I agree… It would depend on the case and how many supporting reads there are by position.
*Thread Reply:* But 2 is very low, it's a 63% base miscalling probability.
*Thread Reply:* I know, but if you check, several papers from CPH have used this parameter
*Thread Reply:* @Åshild (Ash) do you happen to have any insight on that (I know you don't really work in those groups, but maybe you've heard/been in discussions?)
*Thread Reply:* I think it is used because of something related to the error probability of base calling with the older Illumina HiSeqs, and that that was a parameter used by Illumina. But I’m not entirely sure.
Have any UK-based people had a hard time getting KAPA HiFi HotStart ready mix? Do any UK-based suppliers sell it?
*Thread Reply:* Is there any lab-based (or otherwise) product that UK is not short on currently?
*Thread Reply:* lol fair. but I can’t even find a provider that’s “out of stock”
*Thread Reply:* If anyone ever comes across this post with the same question, I could not find a way to order it without directly contacting Roche and getting a quote and placing a sales order. Farzad is the man and can help you meet all of your KAPA needs: farzad.javad@roche.com
Hello experts! Would it theoretically be possible to identify the geographic origin of plant remains recovered from dental calculus (let's say Medieval/post-medieval) from their DNA? And what would the practical challenges be? (I'm assuming fragmented DNA and the overwhelming oral signal would cause issues?)
*Thread Reply:* Theoretically yes.
You would need sufficient DNA reads/coverage on loci on the plant genome that you can do populatio ngenetics - assuming the given species has suffcient structure diverstiy that it can be linked with location
*Thread Reply:* Primary issue would indeed be overwhelming oral signal meaning it's difficult to retrieve the plant reads
*Thread Reply:* Theoretically is all I need for a grant application 😛 well, some grants...

*Thread Reply:* I mean theoretically yes, but a lot of plants have enormous genomes with lots of repetitive elements, so without a huge amount of plant reads/a very successful capture, it will be... Challenging. To say the least.
*Thread Reply:* Yeeeeeaa thought so... Any insight on protein-markers that could be useful @Zandra Fagernäs?
*Thread Reply:* Wouldn't you need a "genetic atlas" of plants to project your sample onto? I mean, in human genetics you can predict ancestry of an individual e.g. based on a PCA on genotypes from thousands of other human individuals with known ancestry
*Thread Reply:* That's what I mean 'on loci on the plant genome that you can do pop-gen'
Hi, all. Does anyone know why the Meyer and Kircher 2010 blunt end direct ligation protocol is used preferentially with aDNA? Does anyone do blunt end lib preps with a-tailing and Y-shaped adapters with aDNA? Is blunt end direct ligation more efficient?
I think the Orlando group may have experimented with this a bit: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0078575 (I don't think that directly addresses your question though)
Yes, many groups had serious problems with aDNA and TA ligation. Blunt end ligation was found to work better empirically
*Thread Reply:* Do you know of any other papers that describe that?
*Thread Reply:* Or is the Orlando one the main one?
*Thread Reply:* Thats the published one
*Thread Reply:* Has somebody tried the Santa Cruz Reaction Protocol?
*Thread Reply:* @Katerina Guschanski maybe ask on #general?
*Thread Reply:* Thanks so much, @James Fellows Yates and @Christina Warinner! This is good to know. I imagined there must be a reason why blunt end ligation is used almost exclusively in aDNA despite AT ligation being the mainstream method in genomics.
*Thread Reply:* @Katerina Guschanski Yes, we (Tom Gilbert’s group) switched to using SCR for all aDNA work in 2020, it works really well in my experience 🙂
*Thread Reply:* Brilliant! Any chance you have a protocol you'd be open to share? We'd like to test it in comparison to our standard double-barcode double-index protocol
*Thread Reply:* There is a full protocol supplied by the authors in their supplementary material https://oup.silverchair-cdn.com/oup/backfile/Contentpublic/Journal/jhered/112/3/10.1093jheredesab012/1/esab012supplsupplementarymaterial.pdf?Expires=1649091871&Signature=QTyxLLQOb1QgZ-YTCdBng8xd9rC3rZul8qX7-dUD18KIFRN~GjkQyemPD0mRfv11fcwJQTjfxl9su8u8XvdC-xUKDf5C5fv8X5xi~D7tY0pW04NZNcZh5dhrEmo46Oc0Oc3nMQ0hvm0VqvfGAq9qmPkHccrQKtgzdtBPghfMBHcfsWYToYRAOq2OilKaQ8dCUJ0gm9ewLL9wh4vrrperSEKxdTaCe~amI-K-Vo0fD0HFN8EN32K7ecx1qD8M~ngq68dm73tAI9rt6ZzMPZzZ6nNeJC-xDI5o~ZbtlJ2DGPIyEjSoOhwNkIeGjBnb0gpWls-rtdg6NdyCNHg77r-X4A&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA|https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jhered/112/3/10.1093[…]ls-rtdg6NdyCNHg77r-X4A&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA which is the same as the one we use 😉
*Thread Reply:* That's rare that a protocol from the paper can be taken and applied without tweaks 🤪
*Thread Reply:* Thanks a lot, @Åshild (Ash)
*Thread Reply:* The only major difference is that we do a SPRI bead clean-up after the library build instead of MinElute cleanup.
*Thread Reply:* Any specific reason why?
*Thread Reply:* it’s faster and less pipetting
*Thread Reply:* Hahaha, true that!
*Thread Reply:* The protocol they provide in the paper is really good and explains the steps well , it’s true that many of the other protocol papers out there are often lacking in detailed explanations.
*Thread Reply:* That's really comforting to know
*Thread Reply:* We had issues with the Santa Cruz protocol. We got a lot of primer dimmers. @Maria Lopopolo or @Miren Iraeta Orbegozo (@Iraeta Miren) can give more details about the issues we had, as it was them that did it. @Betsy Nelson on the other hand has just spent some time in California learning the protocol and may be able to give some details on the tricks to make it work. But for us, our first shot was not very good.
*Thread Reply:* @Nico Rascovan did you QC the adapter and splint oligos before using them?
*Thread Reply:* we QC’ d the splits and adapters. I think this library prep success it’s really dependant upon the type of sample and how you can fine tune beads clean-up and the c1-c5 dilutions e.g. more stringent beads to DNA ration post adapter libration or adding less adapters (e.g. one step below suggestion) at ligation step. I think Miren is now testing a few of these adjustments and was seeing less dimers 🙂

@Miren Iraeta Orbegozo has joined the channel

Hi folks — I’m wondering if anyone else has been having trouble (or has solved the issue of) downloading ENA files which give FTP directory errors recently? Trying both ENA’s enaDataGet scripts and wget directly (and across different projects), I sometimes get Error with FTP transfer: <urlopen error ftp error: error_perm('550 Failed to change directory.')>. Googling suggests that this often happens when the directory doesn’t exist or perhaps in the case of permissions issues, which is not something I could resolve on my own ofc 😕
*Thread Reply:* Hey Olivia, can you post a few example commands to see if we can replicate this?
*Thread Reply:* The ENA servers are sometimes a bit shaky, so it could just be a connection issue - particularly if you're trying to download from the US.
*Thread Reply:* You could consider trying to download stuff with pipelines such as nf-core/fetchNGS which does some retrying mechanisms for you.
Or even try: https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump
which should be able to download ENA data but from the SRA mirror of the ENA (as SRA/ENA basically mirror each other and has the same data)
*Thread Reply:* Ooh thank you for the tool recommendations, those are good to have and I’ll check them out! The good news is that I had a labmate test it and I also tried it locally, both of which succeeded, so ENA isn’t the issue. The bad news is that my profile on our advanced computing cluster is still being rejected! But that’s a problem for our computing team, so I’ll leave it at that. /endthread
Hey all, wondering if anyone has got experience working with sourcepredict, and might be able to help me? Just keep getting an error and I don't know what I'm doing wrong.
It seems like step 1 runs smoothly and it tell me something like:
- Sample: 7309-02minikrakenAll_report known:98.61% unknown:1.39%
But then it tries to run step 2, and it keeps saying: Traceback (most recent call last): File "/home/vanme090/.local/bin/sourcepredict", line 8, in <module> sys.exit(main()) File "/home/vanme090/.local/lib/python3.8/site-packages/sourcepredict/main.py", line 172, in main sm.computedistance(distancemethod=distancemethod, rank=RANK) File "/home/vanme090/.local/lib/python3.8/site-packages/sourcepredict/sourcepredictlib/ml.py", line 260, in computedistance tree = ncbi.gettopology( File "/home/vanme090/.local/lib/python3.8/site-packages/ete3/ncbitaxonomy/ncbiquery.py", line 463, in get_topology root = elem2node[1] KeyError: 1
I've tried different Python packages, but they all give me the same error. To get the taxid sample files, I run kraken with (tried both the standard and the minikrakenv28GB databases). It looks like ete3 can't get the ncbi taxonomy, and therefore it also crashes further down the line? Anyone got any ideas what I'm missing/doing wrong?

Hello all, I am looking to test for positive selection using the branch-site model in codeml (PAML), but I am a bit confused with some parameters in the control file (I've understood the main ones including NSsites and model but I am unsure about some of the others). I was wondering if anyone had experience with this, and if they would be happy to have a quick chat or message exchange to confirm my understanding? Thank you!
*Thread Reply:* ooofff, not heard of that tool unfortunately...
@aidanva @Arthur Kocher @Meriam Guellil? I guess you guys work with phylogenies relatively regularly... any ideas?
*Thread Reply:* Sorry have never used that tool either 😶
*Thread Reply:* Thank you all for you replies, and no worries 🙂 I will go by trial and error and see where it leads 😊

*Thread Reply:* Hi Ophelie! I think that Antony had worked with PAML before

Hi all! has anyone here worked with Metaphlan3? I just analysed a sample but got no classification at all: ```#SampleID Metaphlan_Analysis
cladename NCBItaxid relativeabundance additional_species
UNKNOWN -1 100.0``` but I got classification results with Metaphlan2, has anyone had this same issue?
*Thread Reply:* I know @Alex Hübner
*Thread Reply:* Yes, I had some sediment samples that still had something like five species for MP2 but no species in MP3. They completely changed the marker-gene database and there are some species that were dropped during this process.
*Thread Reply:* oh really…. that doesn’t sound good at all… It’s weird though that in my case M2 reported some kind of common fungi but M3 didn’t detect anything… let me show you a snippet:
#SampleID Metaphlan2_Analysis
k__Eukaryota 78.741
k__Bacteria 21.259
k__Eukaryota|p__Ascomycota 78.5878
k__Bacteria|p__Proteobacteria 19.33983
k__Bacteria|p__Actinobacteria 1.91918
k__Eukaryota|p__Apicomplexa 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes 67.16866
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria 19.33983
k__Eukaryota|p__Ascomycota|c__Schizosaccharomycetes 7.90379
k__Eukaryota|p__Ascomycota|c__Sordariomycetes 3.51535
k__Bacteria|p__Actinobacteria|c__Actinobacteria 1.91918
k__Eukaryota|p__Apicomplexa|c__Coccidia 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales 66.18799
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria|o__Rhizobiales 19.33983
k__Eukaryota|p__Ascomycota|c__Schizosaccharomycetes|o__Schizosaccharomycetales 7.90379
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Hypocreales 2.03864
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales 1.91918
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Sordariales 1.4767
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales 0.98067
k__Eukaryota|p__Apicomplexa|c__Coccidia|o__Eucoccidiorida 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Aspergillaceae 66.10824
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria|o__Rhizobiales|f__Bradyrhizobiaceae 19.33983
k__Eukaryota|p__Ascomycota|c__Schizosaccharomycetes|o__Schizosaccharomycetales|f__Schizosaccharomycetaceae 7.90379
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Hypocreales|f__Nectriaceae 2.03864
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Sordariales|f__Chaetomiaceae 1.4767
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Pseudonocardiaceae 1.25179
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales|f__Ajellomycetaceae 0.74471
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Propionibacteriaceae 0.66739
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales|f__Onygenales_noname 0.23595
k__Eukaryota|p__Apicomplexa|c__Coccidia|o__Eucoccidiorida|f__Eimeriidae 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Trichocomaceae 0.07975
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Aspergillaceae|g__Aspergillaceae_unclassified 66.10824
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria|o__Rhizobiales|f__Bradyrhizobiaceae|g__Bradyrhizobium 19.33983
k__Eukaryota|p__Ascomycota|c__Schizosaccharomycetes|o__Schizosaccharomycetales|f__Schizosaccharomycetaceae|g__Schizosaccharomyces 7.90379
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Hypocreales|f__Nectriaceae|g__Fusarium 2.03864
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Sordariales|f__Chaetomiaceae|g__Chaetomiaceae_unclassified 1.4767
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Pseudonocardiaceae|g__Saccharopolyspora 1.25179
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales|f__Ajellomycetaceae|g__Ajellomyces 0.74471
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Propionibacteriaceae|g__Propionibacterium 0.66739
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales|f__Onygenales_noname|g__Onygenales_noname_unclassified 0.23595
k__Eukaryota|p__Apicomplexa|c__Coccidia|o__Eucoccidiorida|f__Eimeriidae|g__Eimeria 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Trichocomaceae|g__Talaromyces 0.07975
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria|o__Rhizobiales|f__Bradyrhizobiaceae|g__Bradyrhizobium|s__Bradyrhizobium_sp_DFCI_1 19.33983
k__Eukaryota|p__Ascomycota|c__Schizosaccharomycetes|o__Schizosaccharomycetales|f__Schizosaccharomycetaceae|g__Schizosaccharomyces|s__Schizosaccharomyces_unclassified 7.90379
k__Eukaryota|p__Ascomycota|c__Sordariomycetes|o__Hypocreales|f__Nectriaceae|g__Fusarium|s__Fusarium_unclassified 2.03864
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Pseudonocardiaceae|g__Saccharopolyspora|s__Saccharopolyspora_unclassified 1.25179
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Onygenales|f__Ajellomycetaceae|g__Ajellomyces|s__Ajellomyces_unclassified 0.74471
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Propionibacteriaceae|g__Propionibacterium|s__Propionibacterium_acnes 0.66739
k__Eukaryota|p__Apicomplexa|c__Coccidia|o__Eucoccidiorida|f__Eimeriidae|g__Eimeria|s__Eimeria_tenella 0.1532
k__Eukaryota|p__Ascomycota|c__Eurotiomycetes|o__Eurotiales|f__Trichocomaceae|g__Talaromyces|s__Talaromyces_unclassified 0.07975
k__Bacteria|p__Proteobacteria|c__Alphaproteobacteria|o__Rhizobiales|f__Bradyrhizobiaceae|g__Bradyrhizobium|s__Bradyrhizobium_sp_DFCI_1|t__GCF_000465325 19.33983
k__Bacteria|p__Actinobacteria|c__Actinobacteria|o__Actinomycetales|f__Propionibacteriaceae|g__Propionibacterium|s__Propionibacterium_acnes|t__Propionibacterium_acnes_unclassified 0.66739
k__Eukaryota|p__Apicomplexa|c__Coccidia|o__Eucoccidiorida|f__Eimeriidae|g__Eimeria|s__Eimeria_tenella|t__GCA_000002835 0.1532
*Thread Reply:* I am not familiar about the changes with respect to the eukaryotes because I don’t use MetaPhlAn for this purpose. However, you have a lot of unclassified assignments and I also saw that I more or less lost all species in MP3 that were named unclassified in MP2.
In the end, it’s up to you to decide what you want to gain from this analysis. MP3 has a higher number of species-specific marker genes, but this might lead to be less sensitive of picking up traces of species. Kraken2 might be a good alternative for this.
There has been recently a new preprint that evaluates the databases for both programs. I haven’t read it myself yet, but it might give some inspiration: https://twitter.com/RobynJWright/status/1519826469610041344?t=e14DGzJS87jjE6EWY7fpOQ&s=09
 }
}
*Thread Reply:* awesome, thanks for sharing @Alex Hübner 🙂
Can anyone recommend shotgun metagenomes for lab air and/or lab surfaces (not necessarily an ultra-clean lab) that I can use to run through sourcetracker?
*Thread Reply:* Hi Bjorn, we used Salter et al. 2014 for this purpose (Accession numbers ERR584320 ERR584333 ERR584341 ERR584348)

Hi everyone ! I will perform a metagenomic analysis on several individuals already sequenced for demographic studies in my lab. What threshold should I use to create the sample list? We thought about only keeping the samples that were 1% endogenous human because this is the threshold we use to decide to sequence more or not in general. I used a threshold of 1X depth human for former projects and we realised that that threshold was too high but I fear that 1% endogenous human might be too low to be able to detect the actual presence of, say, a pathogen.
*Thread Reply:* What type of metagenomic analysis do you want to do? For pathogen detection, generally speaking I don't orient myself on human DNA content. You can have horrible human genome coverage but still get a good pathogen hit. Depends on the sample. But maybe others have used similar thresholds before?
*Thread Reply:* Hey Meriam! I think that for the moment I will focus on pathogens because I don’t really know how to analyse a microbiota yet
*Thread Reply:* Does it mean that when you look for pathogens, you analyse even the individuals that were just screened ? May I ask, what threshold do you use then to know when to capture or sequence more of the pathogen you believe is in there ? Or what would be a good pathogen hit ? (Sorry for the basic questions)
*Thread Reply:* If you just want to look for pathogen I would just have a look at all the samples you have frankly. As for the likelihood of detection it depends on the sequencing depth and the organism
*Thread Reply:* So far we have used KrakenUniq as a first step in our pipeline with a threshold of 200 taxReads and 1000 kmers, so I fear that the pathogens in the individuals that were just screened would fall under this threshold because of the low depth
*Thread Reply:* So with KU you actually get a bunch of stats which can inform you on the hit quality. Imposing a set threshold for reads and kmers like this could result in missing lower hits such as viruses.
*Thread Reply:* in the original KU paper they use the kmer/reads ratio and I also normalise by coverage
*Thread Reply:* Kmer/reads ratio sounds good because it accounts for the fact that you don’t have many reads sometimes. And then you normalise by coverage. Sounds good, thank you very much ! I will try to do that 😊
*Thread Reply:* you can check my haemophilus paper from this year if you want to do that
*Thread Reply:* Like @Meriam Guellil said:
*Thread Reply:*

*Thread Reply:* @Meriam Guellil I could see from your presentation at SPAAM last year and your paper that you used E-value = (kmers / reads) ** cov = 0.001 threshold for filtering your KrakenUniq output. Could you please elaborate on why this kind of combination of KU stats and why this E-value threshold?
*Thread Reply:* Hi @Nikolay Oskolkov ! So the kmer/reads ratio is from the original paper of KU and you can expect the reason why it is used is that for a good hit you should have n times more unque kmers than reads. So for example if what you have is mostly overtiling it should technically be represented here. I started testing around in late 2019 (i think...) because I was switching from K2 to KU and I wasn't too happy in using reads or kmers for colouring heatmaps for example because depending on the genome size/structure etc these values or cutoffs for them could be widely different. From the tests I did back then this combination of stats ended up working best for me. Now keep in mind I use it almost exclusively for pathogen detection. So this gives me a nice cut-off value from when hits might be worth investigating even if they are low coverage and also eliminates noise in my heatmaps. The thresholds I have used are variable. 0.001 is usually as low as I go, mainly in order to still catch very low viral hits. There might be better combinations out there though, this is just what I ended up using and for my purposes it has done a great job.
*Thread Reply:* I see, thank you @Meriam Guellil! I can understand the intuition behind constructing a kmers / reads variable for filtering because "kmers" and "reads" are pretty correlated so perhaps one of them can be treated as a redundant, so one could replace the two by a new one which is their ratio. However, I do not fully understand why to multiply this ratio by "cov"? Why multiply and not sum or divide? Also, why "cov" and not "dup"? I just want to understand the intuition behind this combination of KU stats that you used
*Thread Reply:* Like I said I did try a couple of variant including dup but dup is already taken into account with the unique kmer count. The reason i use cov is because while the ration kmer / reads gives you a good indication it doesn't account for how much of the kmer dictionary for the taxon is actually covered, since the kmer numbers can differ and that way I can visualize and filter them irrespective of size
*Thread Reply:* And like I said this is what I came up with but I am sure there a multiple and maybe better ways to pair the stats
*Thread Reply:* Are you using the total kmer count for each taxon, or the unique kmer count for each taxon @Meriam Guellil?
*Thread Reply:* unique kmer count. With total kmer count do you mean for the lower ranks as well?
*Thread Reply:* kmer (including duplicated ones) assigned directly to a taxon
*Thread Reply:* KU lists number of distinct or unique kmers
*Thread Reply:* So unique kmers follow read counts (at least according to KU paper fig 3. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1568-0/figures/3) So shouldn’t K/R be more or less a constant ?
*Thread Reply:* @Maxime Borry this is what I mean when I say that those two are correlated, so if you know one you will find the other one, so no need (perhaps) to filter with respect to both of them, one of them (or their combination such as ratio) is good enough for filtering
*Thread Reply:* But that made me check the definition of “coverage” for K2 vs KU @Meriam Guellil, and that’s actually a different one:
• in KU, they use unique_kmers_for_clade / genome_size (https://github.com/fbreitwieser/krakenuniq/blob/2ac22bf7681223efa17ffba221231c7faac9da05/src/taxdb.hpp#L1107)
• in K2, they don’t provide it, but imply that it would be uniq_kmers_for_clade/total_kmers_for_clade https://github.com/DerrickWood/kraken2/pull/249#issuecomment-638311769
*Thread Reply:* but you can have a high read count while still having a low unique kmer count which is what they point out in the paper, so the ratio for sure makes a difference. you can just use the base ratio it one its own too
*Thread Reply:* Yes I agree with Meriam, normally the good hits have way more kmers than reads and when it’s not the case I doubt more the validity of the hit
*Thread Reply:* what can happen though is that you have a super high coverage genome in a utopic world and you have the whole genome covered multiple times in which case the unque kmer count wouldn't go up anymore but the read count will. So if the cov is 1, then there is no point.
*Thread Reply:* Fortunately, we are not used to work with utopic data 😂 but yes you are right 👍
*Thread Reply:* I see. But in that case, would it make sense to give more emphasis to the coverage ?
Maybe something like E = K/R ** double_exp(C)
This way for:
• low coverage, high duplication: K<<R so K/R -> 0, C~=0 so dodouble_exp(C) ~ 1, then E->0
• low coverage, low duplication: K/R -> ~(read_len - kmer_len), double_exp(C) ~ 1 , E->~(read_len-kmer_len) because you have up to read_len - kmer_len kmers of length kmer_len in a read of length read_len (read_len - kmer_len + 1 to be precise)
• high coverage (up to 1, because KU uses uniq kmer for “coverage” computation), low duplication: K/R -> 1 ,double_exp(C) -> inf , E-> inf
The assumption being that for low coverage, the K/R ratio is what matters, for high coverage, the coverage is what matters
*Thread Reply:* Sounds good! I am/was lacking the mathematical background to go deeper into the rabbit hole I am afraid so happy to see this improved. I will have a look 🙂
*Thread Reply:* Interesting suggestion @Maxime Borry, but why to collapse them into a single variable, why not to filter with respect to each of them separately? The thing is that 2 out of the three variables seem to be orthogonal / independent. Then constructing a single variable out of two orthogonal might reduce the quality of filtering
*Thread Reply:* I mean to filter in 3-dimensional space is probably more intuitive than in 1-dimensional
*Thread Reply:* We can construct different functional combinations out of the 3 KU stats, with different asymptotics, but do we really need to have one single threshold for filtering?
*Thread Reply:* For example the way GATK filters good from poor quality variants, it uses 17 QC metrics and implements different thresholds for each metric. In our case, for example, we can compute distributions of kmers, cov, reads, taxReads and dup and exclude organisms in the tails of the distributions
*Thread Reply:* Next, one can e.g. compute a PCA on the kmers, reads, taxReads, cov and dup stats, where data points in the PCA plot are the oganisms, and one could perhaps detect clusters of organisms with good QC metrics and poor QC metrics. That would be a multivariate filtering approach. Fir example, a PC1 could be a linear combination of the 4-5 KU stats that can be used in case one definitely wants just one single threshold. But why not to filter in 4-5 dimensional space? It is not too many dimensions to tackle
*Thread Reply:* Hmm, that’s another interesting approach
*Thread Reply:* Acutally, just realised that it would need a “double exponential” function for the coverage, because KU cov will be a max of 1 (since it’s using unique kmers). https://en.wikipedia.org/wiki/Double_exponential_function
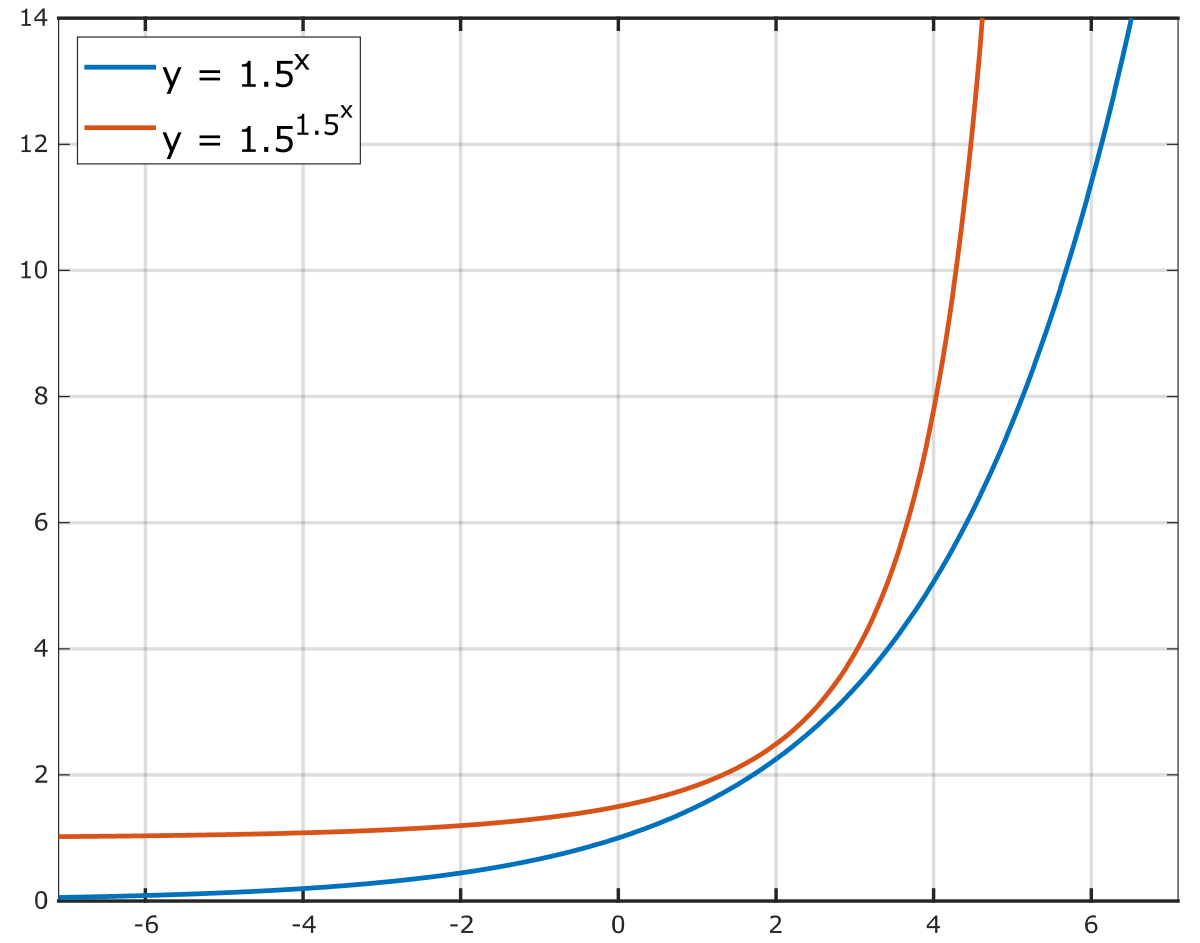
*Thread Reply:* I made a small notebook to compare the effect of adding the double-exp to the coverage
*Thread Reply:* Sorry but I am not sure to understand the final plots @Maxime Borry. Does it mean that Meriam’s e-value is better than the double exponential or the opposite ? I don’t see the lines for case c and d on the penultimate plot and for the last plots, two cases are called b so I’m confused 🙈😅
*Thread Reply:* I made a blog post out of it 😉 Hopefully it’s clearer https://maximeborry.com/post/kraken-uniq/
Hello all, I was wondering if anyone has had experience with setting up Path Sampling for model selection with Beast2? I'm completely lost in how you're meant to alter the xml file (http://www.beast2.org/path-sampling/). I did manage to run a Nested Sampling analysis, but it's quite unstable and keeps crashing, hence why I am thinking of trying the Path Sampling approach. If anyone has any experience, I'm game! Thanks 🙂
Hi all! Is there a list somewhere of bacterial species and whether they are aerobes, anaerobes, etc?
*Thread Reply:* I use the bacdive API to ask for this, but it's a bit fiddly.
*Thread Reply:* Or the bacdive website itself if you only want to check a few manually
*Thread Reply:* https://bacdive.dsmz.de/
*Thread Reply:* There is also a R package called bacdiveR but it may not work anymore (even though it was very good)
*Thread Reply:* You will need to make a consensus across multiple strains of a species in some cases
*Thread Reply:* Thanks! I'll try the API and R package and see how it goes
*Thread Reply:* Can confirm that bacdiveR is currently broken
*Thread Reply:* Thanks, that'll save me some time
*Thread Reply:* Ok, you have to do it more manually then but it is possible still within R, there are various packages for dealing with rest APIs. You just have to wrangle list vectors a lot into tables manually
*Thread Reply:* Any experience with BacDive? It looks like it's only on https://r-forge.r-project.org/R/?group_id=1573 and there's not much other info I can find
*Thread Reply:* https://api.bacdive.dsmz.de/
*Thread Reply:* Oh that's the same thing sorry
*Thread Reply:* That's new, not used it before sorry
*Thread Reply:* But looks good!
*Thread Reply:* At least it looks like it's being actively maintained
*Thread Reply:* Maybe you could write a little tutorial if you get it to work
*Thread Reply:* I would be curious to know how it works
*Thread Reply:* Sure, that'll force me to document my steps properly 😅
*Thread Reply:* Could even put it on the <#C02D3DJP3MY|spaam-blog>
*Thread Reply:* I think others would be interested to know how it works
*Thread Reply:* I assumed that was the reason you asked? 😉
*Thread Reply:* @Ele @Shreya ☝️
*Thread Reply:* But I'm genuinely curious, there is a little project I want to do when I have time (ahahahahaha) which getting such info would be really important
*Thread Reply:* So if there is a good way of doing that it would be really useful to know
*Thread Reply:* What's this 'time' you talk about? 😆
*Thread Reply:* I think you can find it at a place called Atlantis
*Thread Reply:* It's made by a people called mermaids
*Thread Reply:* There’s also a working BacDive API client for Python 🙂 https://pypi.org/project/bacdive/

*Thread Reply:* Looks pretty similar to the R package, although I like the ability to filter! If only I knew python... 😅
Hey all! I've been doing some work on ancient pathogen detection and have been using several authenticity criteria (fragment length, damage pattern, evenness of coverage, & edit distance). I'm now looking at some oral microbiome species, and it looks like I'm getting some complete genomes (very excited!). I just wanted to check with you all what you use as authenticity criteria. I kind of figured that they would be the same?
But for example, I don't see edit distance and evenness of coverage mentioned much in the oral microbiome literature, but this is still important I guess? I've got a couple of perhaps ambiguous ones that I'm not sure about whether this is real. I've attached some examples. MS10211A.bacterium looks real to me. But what about 7401-03A.oral? Edit distance looks good, but evenness of coverage not so much. And MS10211_A.oral? I'm using bwa aln for the mapping with seeding disabled, -n (error-rate) 0.1, and gaps open 2.



*Thread Reply:* Generally it's the same concept yes, but with some additional things (I apologise if you know some/all of this already, but I'm addressing common misconceptions by aDNA researchers)
However you have to be careful: you can't really equate pathogens to standard commensal/pathobiont species, particularly when it comes to the oral microbiome.
General concepts are: what you see reported as 'disease' causing oral taxa are often from clinical studies that only look specifically at that taxa. In fact modern oral microbiology is showing very much that most of these 'disease-causing' taxa are always present in the oral cavity even in health individuals, it's just when the equilibrium is broken disease happens (i.e. something disrupts the relationships causing some taxa to 'take advantage' and over-grow or do other things). So you have to be EXTREMELY careful if you want to report this from a disease aspect (just saying as you're coming from a pathogen direction in your earlier stuff).
Secondly, you must be extremely careful how you are reporting your hits if they are ACTUALLY the species you've mapped against. The oral microbiome is extremely rich (>= 700 species detected, and estimated 100-300 taxa at any one point in western individuals [who have reduced diversity somewhat]). You will have many strains or very closely-related taxa of species. So this means when you're mapping against a reference genome, you could be picking up lots of reads from closely related strains etc, which means if you do variant calling you may accidently incorporate multi-allelic positions and you'll call up the incorrect variant (edit distance doesn't help here as you're dealing with closely related strains so you don't necessarily expect multiple variants on a read). You may also pick up genes present in the reference genome (which are actually often weird clinical variants that don't represent a 'typical' representative of a species). So you if you want to do phylogenomic analysis you must be very careful to check you're not making a wierd 'chimeria' genomes that may bias your mutation rates/reference bias etc. I tried to address this in my PNAS paper from last year but didn't find a good solution (although it was only a small part of the paper).
If you use a genotyper you can sometimes pull multi-allelic SNPs out of your VCF file and display this. IIRC Warinner 2017 (Annu Rev. Human. Gen) describes this theortically, and Vagene 2018 (Nature Eco Evo) has an example of this using MultiVCFAnalyzer output (but you don't have to use that).
*Thread Reply:* \lecture (sorry)
*Thread Reply:* I very much agree with James, but can add that I would highly value evenness of coverage and edit distance despite they are indeed sort of under-emphasized in literature. Your plots look like true hits to me although as James said I would be careful interpreting them as pathogens. And evenness of coverage and edit distance are handy to filter out majority of false-positive hits but sometimes they are not enough either. A typical situation when detecting microbes is that reads from a species A can happily map to species' B reference genome even if those two are not close enough. Then, evenness of coverage and edit distance (and also deamination pattern to some extent) can help you to figure out that you are probably mapping your reads to a very wrong reference genome. However, when there is some resemblance between A and B, and you work with deeply sequenced samples, you might get a descent coverage of species' B reference genome by reads originating from species A. In this case you probably need some phylogenomic insights. Namely, if you happen to know species-specific alleles that are present only for your organism of interest and absent in all other species from the corresponding genus / family, this would be a way to separate the reads that truly originate from your organism of interest from the reads originating from closely related species. However, to establish species-specific alleles for your organism of interest, you need population level allele frequencies for your organism of interest as well as closely related species, which is a tricky, expensive and rarely feasible thing 😞 On a positive side, it looks to me that you have done a great job with the first detection screen where you found a few good candidates for further validation! 🙂
*Thread Reply:* Yes, agreed evenness of coverage also very useful here (but look reasonable from your plot)! And I also agree first screen looks good!
*Thread Reply:* > . However, to establish species-specific alleles for your organism of interest, you need population level allele frequencies for your organism of interest as well as closely related species, which is a tricky, expensive and rarely feasible thing Yes this is very important. The problem (Which I forgot in my essay) is that most oral microbita are quite undercharacterised. For example for Tannerella, up until 2018 had a single published genome, and then this was expanded to 8 and now something like 15 in the space of of 4 years. Very few oral taxa have more than a single genome and often are not even named! So phylogenetic analyses are difficult to do.
So you just have to be take a lot of care :)
Hi All!
We were wondering today if anyone out there could provide some feedback, or point to specific papers, on performing functional enrichment tests on sets of genes within a meta-community?
Starting with abundance estimates for microbial metabolic pathways and other molecular functions from our shotgun sequencing data (HUMAnN), one would typically perform some kind of association test on these functions to determine which ones are more strongly associated with one group of samples over another.
But we are considering if enrichment tests would require any additional considerations when performed on a set of genes within a metacommunity. The hierarchical structure of these tests could provide a broader look at functional enrichment across the community compared to just considering abundances under a single pathway.
Any thoughts?
*Thread Reply:* @irinavelsko any ideas? 🤔 Or someone from OU?

*Thread Reply:* I don’t quite understand the question, but here’s how I think about functional data for my analyses-
*Thread Reply:* For HUMAnN analysis, I don’t consider the species-level assignments for the first step. Instead of looking at a metataxonomic community, I think of it as a metagenetic community (a community of genes/pathways, independent of the organisms they come from)
*Thread Reply:* I perform the same kind of enrichment tests and statistical tests on a gene or pathway matrix as I would on an OTU table, b/c just substitute your-favorite-taxonomic-level for genes or pathways
*Thread Reply:* then once I have an idea of what’s enriched in one group or another, I look at the species that the genes/pathways are coming from
*Thread Reply:* I don’t think with aDNA we can get the level of resolution on species contributions to pathways/genes that are needed to dig into species-specific contributions
*Thread Reply:* Also all bacterial genomes are full of unannotated proteins, so we’re missing a lot of information simply b/c we can’t say what all genes are or what pathways they’re involved in and what the products are
*Thread Reply:* I think the HUMAnN paper actually uses simple statistical tests of enrichment, which are built in to the program as separate scripts, which was acceptable at the time
*Thread Reply:* Do you have a specific question though?
*Thread Reply:* I would look for papers that have used HUMAnN to get an idea of what people in modern metagenomics are doing, if you’re looking for creative and interesting approaches
*Thread Reply:* Sorry just returning to this thread now. Thanks for the feedback so far @irinavelsko!
My question was more specifically tied to gene set enrichment tests which would be more commonly applied in differential expression or population genomics studies. Sure, the methods commonly used with HUMAnN output with give you what pathways/genes are "enriched", but when it comes to the enrichment of a function in this type of data (you could say the combined enrichment of multiple genes/pathways under a GO term) we were thinking of applying methods used in packages like topGO, which will do something like a Fisher's exact test on a set of genes that are mapped to GO terms, while keeping the hierarchy of GO terms.
Hopefully the question is clear, it is a bit off the wall though, and I haven't found papers that address this specifically 🤪
*Thread Reply:* Haven't tried it yet on aDNA, but from my time working on transcriptomics, I had a good experience with eGSEA
*Thread Reply:* https://bioconductor.org/packages/release/bioc/html/EGSEA.html
*Thread Reply:* If I understood correctly, that's GSEA (Gene Set Enrichment Analysis) that you're looking for
*Thread Reply:* All these GSEA methods usually start with a list of differentialy expressed genes (DEGs) that you get from one of the differential abundance method (edgeR, DeSeq2, limma, ...) and then look for these in pathways/gene-sets.
*Thread Reply:* eGSEA is nice because it combines different GSEA methods and gives you a consensus of their results
*Thread Reply:* That sounds neat! I've used ReviGO (http://revigo.irb.hr/) but I don't find GO terms useful for bacterial functional analysis and prefer other gene classification systems
Hi All, For everyone using Kraken2, have you tried building databases with differing k-mer lengths and tested how this changes the number of reads assigned taxonomy, and how accurate the assignments are?
I have some libraries with lots of very short reads that had very few reads assigned taxonomy using a Kraken2 database built with the default k-mer length. I’m considering trying to build a database with a shorter k-mer length, but thinking that it might not be worth it if the assignments are too non-specific.
Does anyone have any experience with this they can share?
Hello Irina. Are your reads shorter than 31bp?
Hi @irinavelsko, I never tried to use kmers shorter than ~30 bp since they are too non-specific as you say. However, once I tried to do alignments of ultra-short reads (the vast majority were shorter than 30 bp) against a large (NT) database, and got approximately 1-2% of reads aligned with MAPQ>0. So I believe it might make sense to build Kraken db with kmer length shorter than 30 and run classification of ultra-short reads but my guess would be that it will not help you much, i.e. I believe very few reads will be classified and even fewer will be accurately classified
Hi Nico and Nikolay, here’s an example of the read length distribution from a Damageprofiler run against a highly abundant species in one of the samples. Most of the reads are >30pb, and the average length is ~45bp. Many of the samples look like this for each of the species mapped against for Damageprofiler, and they had ~1-2% of reads assigned taxonomy by Kraken2

ok, super. Correct me if I am wrong, but all reads with length >31 should be classified with the default k-mer size of kraken2. Maybe you can just run some tests only of the reads below 30bp, but I agree with Nikolay that such reads are more prone to misclassification. Depending on how many they are, you can probably Blast only those against NCBI nt, then import to MEGAN and see how they look like
based on your distributions it seems that there are just some thousand reads with length 25-30bp
Maybe centrifuge would be interesting for you since it has extending kmers but I agree that I wouldn't trust reads shorter than 30bp too much
The mode of the length distributions seems to be around 30-35 bp, so I would not bother making a new Kraken DB (which takes a lot of time if your database is large enough) to classify / rescue additional a few thousand reads
I think is not so much about whether you can assign very short reads. The question would be rather whether you have a different sensitivity of assigning reads to taxonomic units if you use a k-mer length of 30, 35, or 40 or any other number. The longer the k-mers, the more specific they are, but due to the shorter reads we cannot use very longer k-mers. So if you have a read of 45 bp length, then you will be in theory able to assign them with all three k-mer lengths. Did someone ever did test the difference in specificity per k-mer length?
@Alex Hübner I agree, sensitivity vs. specificity in terms of e.g. ROC-curve is an interesting question, which can be computed on simulated data where one knows the ground truth. We are soon going to publish our ancient metagenomics workflow that I briefly presented last SPAAM, and for this manuscript I have done some benchmarking of available workflows (classification + alignment) on simulated data in terms of ROC-curves. However, I have not tested specifically the effect of kmer size on ROC AUC. There can be such benchmarking already done in papers that compare classifiers (CLARK, centrifuge, Kraken, MetaPhlan etc.), there are a few papers on this topic, I believe @irinavelsko knows classifier comparison studies better than me. My intuition here is the following. Since our reads are relatively short, I would indeed not select very long kmers (like 40) when building a database because then we will loose lots of reads, so we might be loosing sensitivity. On the other hand, kmers shorter than 30 might be very non-specific, so we jeopardize specificity. So for a rough sensitivity-specificity balance, I would select the shortest "specific-enough" kmer size, which seems to be around 30, therefore I believe this is the default in Kraken. But this is just my intuition, I haven't properly tested this
Thanks for the detailed answer @Nikolay Oskolkov!
Last march we have published a benchmark study “Benchmarking metagenomics classifiers on ancient viral DNA: a simulation study” (https://peerj.com/articles/12784/) where in a supplement note https://doi.org/10.7717/peerj.12784/supp-9 we have investigated the outcome of a Kraken2 database with a smaller kmer. You can classify more viruses, but the error rate increases also significantly.

Thanks @Samuel Neuenschwander, that’s what we were looking for. It’s impressive to see how many more spurious taxa you got when dropping the k-mer lengths from 35 to 29 bp in Kraken2.

Just to chime in here, @irinavelsko and @Alex Hübner. We have explored different kmer sizes with Kraken (original) and our ancient mammalian mtDNA captures. We've essentially found that a kmer of 21 is a good compromise of true to false positives, with the caveat that we are then mapping to mtDNA genomes, and taking only reads that have a good MQ and are at least 35bp long. The mapping process itself removes a lot of the false assignments you get with Kraken, and at higher kmer sizes we fail to assign a lot of reads. We've tested this with simulated ancient DNA, but none of this is published yet.
On the subject of the correct kmer size - I'm curious about the thought "my average read length is 40, so a kmer of 30 will be ok." With those kmer/read sizes, each read has only a handful of kmers - and any mutation/damage/error in the middle of a read will disrupt all possible kmers in that read, right?
And thanks for the paper link, that indeed looks like a good reference!
Hi! I am trying to learn how to do qpWave/qpAdm analysis through Admixtools command line (I am having a hell of a time trying to get admixr/admixtools2 to install on R to no success...) and want to work my way through this tutorial: https://comppopgenworkshop2019.readthedocs.io/en/latest/contents/05_qpwave_qpadm/qpwave_qpadm.html. Does anyone know where the data comes from? It does not appear to be the same data as that used in the admixtools example data/Patterson 2012. Thanks a lot!
*Thread Reply:* Hi Audrey, are you applying this to microbial data or something? I thought qpwave stuff was designed for eukaryotic genomes and pop-gen
*Thread Reply:* Yeah I am trying to apply it to eukaryotic data, I apologise that it is not appropriate for this channel but was hoping someone might know and one of my colleagues had suggested that I reach out here
*Thread Reply:* I see. Well there might be a couple of people who know but as the purpose of spaam is for metagenomics, and not pop-gen, it's unlikely we have the expertise here sadly :/.
Of course we can keep the question up in case someone does know, but I just wanted to say don't expect much on such a topic here unfortunately. (I keep telling my colleagues in pop-gen to set up their own thing like spaam for your discipline, as we frequently get similar questions etc. but no one has done so yet :/)
Sorry for the slightly negative answer :(
Thinking about it @Marcel Keller has been drifting into pop-gen, maybe he has an idea?
*Thread Reply:* No worries, thank you for your input anyway, and sorry again for the intrusion! I actually am working on a metagenomics project as well and will definitely be back soon with more relevant questions ^_^

*Thread Reply:* Hey @Audrey Lin! You can find all the populations they use in the tutorial in the AARD 1240K + HO dataset here - https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data , so you could download these and use them in place of popgen_qpAdm_test_190120.geno , .snp, .ind. The population names might have changed a bit, so you could either download the version from 2019 to match the individual/population names from the tutorial or edit the tutorial scripts to match the currently used population names.
*Thread Reply:* For instance, the individuals for "YamnayaSamara" in the tutorial are now labeled as "RussiaSamaraEBAYamnaya"
*Thread Reply:* Thank you @Hannah Moots! I guess there is more diversity of people here than I expected 😬
*Thread Reply:* Thanks for stepping in @Hannah Moots ☺️
*Thread Reply:* Happy to! I have worked through the same tutorial myself and am happy if that experience is useful to others :)
*Thread Reply:* And excited to be part of this slack group (just joined recently) and to learn more about ancient metagenomics from you all 🎉
Hi, all. Has anyone had persistent dimer problems with the Meyer and Kircher 2010 DS libraries with dual indexing? If so, what do you think caused it? I haven't before, but I've been working in a new lab and many of my libraries have dimer peaks. I know low starting template can lead to this, but I haven't really had dimer problems with this protocol before even in blanks. Thank you!

*Thread Reply:* Yes! Occasionally with low input samples, but mostly with material that tends to cross-linking/PCR inhibition like fixed tissues or some plant species. Trying a bead cleanup with different library to beads ratios sometimes gets rid of it
*Thread Reply:* Also worth checking if the adapter and index stocks are at the same concentration as your previous lab 🙂
*Thread Reply:* Thank you! These libraries are from Neolithic and Mesolithic skeletal remains, so they're quite old and preservation is not great. But I'm just curious why I've never had issues with dimers even in blanks before, but now they're all flooded. Yes - all stocks and working dilution concentrations are what they should be.
*Thread Reply:* I get dimers in most of my blanks 🤔 Do the libraries look good if not for the dimers?
*Thread Reply:* Here is how I am used to my library blanks (LB) and libraries performing after indexing PCR with no size selection, only column cleanup after amplification. For these I used Pfu Turbo Cx for indexing.


*Thread Reply:* And here are examples of what I am generating now, extraction blank and sample library (this tapestation is old and has some systemic issue which is why the runs are so noisy…they’re all like that). These were indexed with KAPA HiFi Uracil+, but other than that and the oligos being from different suppliers, there are no differences in the protocols or reagents.


*Thread Reply:* I think this would be a good candidate for a 1:1 bead clean up
*Thread Reply:* I have heard that people use 1.3:1 and 1.5:1 ratios as well for ancient DNA. Do you have good success with 1:1? I am worried about losing short fragments in the 170-200 range.
*Thread Reply:* I have excellent results with cleaning up single-stranded libraries with 1:1 and I'm going to try it soon on some M&K libraries that came out with lots of dimers (at 1.7:1). You have a good amount of library compared to the dimer peak and a good size difference. If you are worried about losing short fragments you could try starting at 1:5 and decrease if you don't get rid of it?

*Thread Reply:* Hi Kelly, we used to have a similar effect in post-indexing amplifications of samples or captures with low input. We used to call this effect the “iguana” 🙂. Not sure if what you are seeing here is the exact same thing, but at the time (back in 2014) we were indexing with Pfu Turbo Cx and amplifying (post-indexing) with AccuPrime. Our solution was to switch the AccuPrime with Herculase. I believe the effect was completely gone after the switch. Hope this somehow helps!
*Thread Reply:* Thank you both so much! This has been immensely helpful. I did a 1.3:1 size selection using NucleoMag beads, and it worked so well. The dimer signatures are completely gone, and there wasn’t that much DNA loss or change to fragment size distribution in the 150bp+ range.
*Thread Reply:* Maria, I think that is exactly what is happening, since I haven’t used this enzyme before for indexing. This never happened with Pfu Turbo Cx. I usually don’t re-amplify after indexing unless I have to. When I have re-amplified, I used Pfu Turbo Cx and had no dimers even on very low yield libraries. I didn’t realize there was such variability in enzyme and dimer formation. Thank you!
*Thread Reply:* Good to know! Maybe I'll try 1.3:1 as well 🙂
Hi all! Does anyone have experience using pydamage v. 0.7? I am using it now, however most of the options for parameters listed on the readthedocs website are not actually available https://pydamage.readthedocs.io/en/0.7/CLI.html. For example, I wanted to filter with -t 0 , however it was not possible. I also could not use the --plot or --group functions with the analyze command. Does anyone know if these options are actually available or am I using an outdated version? or maybe I’m missing some components that need to be installed in my conda environment?
*Thread Reply:* How did you install pyDamage? Was it via conda or pip?
@Maxime Borry @Alex Hübner?
*Thread Reply:* Installed with conda inside a conda env
*Thread Reply:* Which version are you using?
*Thread Reply:* —plot and —group worked for me last time I used pyDamage analyze. But Maxime knows for sure 🙂
*Thread Reply:* Maxime has updated it last week but not the functions that you mentioned.
*Thread Reply:* I just checked and I have also version 0.7 installed and pydamage filter -t 0 pydamage_results.csv works for me.
*Thread Reply:* maybe it is the order in which I am listing the parameters in my command
*Thread Reply:* It works now 🤦 😅
*Thread Reply:* Hey Ashild, just tested it again, it works with version 0.70. Also, side note: 0.7 != 0.70 😉
*Thread Reply:* the order of the parameters doesn’t matter
*Thread Reply:* Yes, but, the parameters have to be after the analyze/filter commands , because pydamage filter -t 0 pydamage_results.csv works, but pydamage -t 0 filter pydamage_results.csv doesn’t. I misunderstood the instructions 😅
*Thread Reply:* You're right @Åshild (Ash), this is because they are parameters of the subfunctions (analyze or filter), but not parameters of the main program.
Hi all! For anyone who’s successfully built a KrakenUniq ‘nt’ database, have you seen this message? 0 sequences mapped to taxa. .. accompanied with some 5GB of errors of the sort Didn't find taxonomy ID mapping for sequence XXXX !! ! I’m restarting the build from the previous step in case some file got corrupted but each step takes so long, I thought I’d try here alongside my own troubleshooting attempts just in case someone has a fix.
For reference I’m using a strategy suggested by @Nikolay Oskolkov ages ago, of using Kraken2 to download the full nt database with all necessary files, and then KrakenUniq to build it. Would love any tips! Thank you!
*Thread Reply:* @Shreya the error seems to have something to do with the NCBI taxonomy that does not match the actual sequences. Yes, when I built full NT KrakenUniq DB I used all the same input files from Kraken2, but just started a building process using krakenuniq-build utility. If this way did not work for you could you perhaps take a screenshot of what files are in your directory where you start the building process? I would like to see what files you are using as input, then perhaps I could be more specific
*Thread Reply:* Thanks so much Nikolay! Here’s the output right now, after I restarted it at the sort step last night:

*Thread Reply:* One thing I’m wondering about is if something might be going awry at the shrink stage-- the full database was some 4TB which seemed extremely unwieldy, but I haven’t found data on how to pick a good size to shrink it to. Thank you in advance for all your help (and for all the advice leading up to this!!)
*Thread Reply:* Hi @Shreya, thank you! What version of KrakenUniq are you using? I am quite sure that the database size reduction was broken in versions 0.6, it never worked for me. In the newewest version 0.7 they say this issue was fixed (if I remember correctly), but I did not use the database reduction with 0.7, so can’t confirm. However, your issue does not seem to be related to database size reduction, step 4 failed since taxonomy did not match the sequences in the library-directory. I can see taxdump in your taxonomy folder, did you download it separately from NCBI or you followed Kraken2 instructions? Important that the ids present in seqid2taxid match the headers present in library.fna in the library-folder. Could you check a few?
*Thread Reply:* @Abby Gancz you also had a similar issue of taxonomy not matching seqids, did you manage to solve it and how?
*Thread Reply:* I am using the newest version, 0.7.3, and it does seem like the database reduction worked, or at least it’s not reporting any errors at that stage!
I downloaded taxdump via the Kraken2 instructions:
/gpfs/data/raghavan-lab/bin/kraken2/kraken2-build --download-library nt --db /scratch/shreya23/KrakenUniq_DBs/FULL_NT_06_23_2022 --use-ftp
/gpfs/data/raghavan-lab/bin/kraken2/kraken2-build --download-taxonomy --db /scratch/shreya23/KrakenUniq_DBs/FULL_NT_06_23_2022 --use-ftp
*Thread Reply:* But… the seqid2taxid.map file is completely empty! I will go check into which stage it is supposed to be filled. The library.fna file is definitely not empty!
*Thread Reply:* Hmm, is taxDB also empty?
*Thread Reply:* Could you also please check that ids from accession2taxid in the taxonomy-folder match the headers from library.fna? Also ids from names.dmp I believe should match the headers
*Thread Reply:* taxDB is not empty! Cross-checking IDs now!
*Thread Reply:* It seems like not everything in accession2taxid is in library.fna. But I think most things in library.fna are in accession2taxid? I haven’t checked this rigorously, just pulled a random selection to check.
*Thread Reply:* I don’t think the names.dmp ID numbers match the headers in library.fna though.
*Thread Reply:* @Shreya let me execute the command lines you posted above to figure out what is going wrong and get back to you
*Thread Reply:* Hi @Shreya I checked how I built the NT krakenuniq database and I am quite sure the seqid2taxid.map should not be empty. Do you remember whether this file was downloaded when you ran
kraken2-build --download-library nt --db DBDIR
or you built this file as a step of krakenuniq-build process?
*Thread Reply:* I do not remember clearly where this file came from, and am currently waiting for some disk space to empty in order to start the downloading and building process in order to reproduce the files in your directory
*Thread Reply:* One thing I did in addition to what you have already done, I actually ran the krakenuniq database building on the top of the Kraken2 DB, so I used
kraken2-build --build --fast-build
first, and then proceeded with krakenuniq-build. So perhaps the seqid2taxid.map was built as a step of kraken2-build. Very complicated. But as soon as I can clean up some space I will start the krakenuniq-build and see whether I get an empty seqid2taxid.map file as you got
*Thread Reply:* I feel bad that you would have to clean up space to help me with this! Cleaning out space is miserable!
I don’t remember seeing the creation of the seqid2taxid.map file, but based on the timestamps, I think it was created as part of the build, not downloaded with --download-library

*Thread Reply:* Might make sense for me to delete these and start from scratch with the downloads if you think that would help! I can try it with the kraken2-build --build --fast-build step this time, can’t hurt to have a kraken2 database as well!
*Thread Reply:* Hi @Shreya, my sincere apologies for the very long silence, I had some updates on this but never had time to write a reply to you. I could reproduce your issue and it indeed looks like the seq2taxid.map file is built incorrectly by KrakenUniq when using the taxonomy downloaded by Kraken2. I have not fully understood the difference but it looks like Kraken2 and KrakenUniq use slightly different taxonomy formats, therefore a taxonomy file downloaded via Kraken2 is not compartible with KrakenUniq way of database building. A workaround, as we discussed, is to construct the seq2taxid.map via Kraken2 database building procedure, and then use the seq2taxid.map for building a KrakenUniq database. I am afraid, I do not have better suggestions right now, but I am going to carefully compare the taxonomies from Kraken2 and KrakenUniq and hope to figure out the difference. Will keep you updated!


Hey all, I'm kind of running into a last-resort problem. I was hoping to use my University's compute cluster to analyze a few hundred genomes in EAGER but it is continually down and broken and even randomly deleting users, and we have no other compute infrastructure.
Does anyone rent out or allow external access to some computing infrastructure I could use to complete my dissertation project? Thanks!
*Thread Reply:* Do you have any budget?
*Thread Reply:* Not a lot to almost none, but I can maybe scrounge up some funds, or assuming it doesn't cost a lot of money I can probably sacrifice out-of-pocket money.
*Thread Reply:* You could maybe consider the cheaper ends of AWS/GCP or something if you don't store your data there too long, but steep learning curve and you can spend a lot if you're not careful (they might have educational offers).
Otherwise I don't really know for the US, but in Germany at least there are a few multi-institution (e.g. gwdg or national initiatives (deNBI is one) to provide 'free' computing to German academics. You just need a German PI. So if you could find a collaborator in Germany that may be one route but maybe there are similar schemes in the US?
You could also maybe see if there are other clusters at your uni in other departments, you might be able to slip into one of those via your supervisor
*Thread Reply:* Thanks, James. I'll look into what I can do with AWS then. We have no other clusters or bioinformatics support on campus
*Thread Reply:* Not even the physicists or something like that?!
*Thread Reply:* There are also other companies that offer more normal large VMs
*Thread Reply:* But I don't know why by name. It may give you a smaller learning curve
*Thread Reply:* Nope, as far as I know the cluster that we recently got funding for and installed that has been breaking down constantly was the first cluster our campus got. Ok I can look into that option as well, thanks!
*Thread Reply:* Oh wow... Ok!
Has anyone looked into the extent of microbe degeneration through mineralisation that would cause us to be unable to detect certain bacteria consistently in dental calculus? In other words, are there bacteria that don't show up in dental calculus simply because they are more prone to mineralisation, and are therefore too degraded to be detected?
*Thread Reply:* Btw, a question like this is better on #general, many more eyes and it's a good question ;)
*Thread Reply:* I think mineralization should help improve their preservation
*Thread Reply:* Yea over the long term I agree, I was just curious about any immediate damage caused by the mineralisation process
*Thread Reply:* Trying to understand why my model calculus is distinct from the growth medium samples I took. The likely explanation is maturation (as in @irinavelsko's 2019 paper), just looking for possible alternate explanations
Could someone jog my memory about studies that link presence/absence of dental calculus deposits in different species with species physiology, tooth morphology, diet, etc.? I know I've seen all this somewhere but struggle to dig up the relevant papers

Hi, I have questions about using Pydamage (@Maxime Borry).
- I know that damage patterns in contigs depend on preservation and environmental conditions. But, from your experience, can you recommend any threshold for C->T transitions below, which is very unlikely to be ancient? e.g. I have a contig with frequency at the first three positions: 0.031, 0.024, 0.01. Values are really low, but conditions are also very good for DNA preservation. Should I believe that it is an ancient fragment?
- I used pydamage filter -t 0 pydamageresults.csv, but still, I observed some obvious lab contamination with C->T frequency at the first three positions: 0.002, 0.001, 0.001. I don't understand why this evident contamination with a very low damage pattern was filtered as an ancient and why predictedaccuracy for them was 1?
- Why coverage from metaspades is different to coverage in pydamage results, even though I used the same dataset for assembly and mapping? Thanks a lot for your help!
*Thread Reply:* Note Maxime is on a conference this week so might be slow to reply
*Thread Reply:* 1. No, and that's part of the reason why we developed PyDamage 😉
- The predicted accuracy is very high because you most likely have a lot of reads mapping on a relatively long contig. So in a way, it tells you that even though the difference is very slight, it is consistently there. But again, there's no hard threshold that would for sure tell ancient from modern DNA, it's all about concurring lines of evidence
- Because you get the coverage from a bam file of the reads realigned to your contigs using a short-read aligner. The way metaSpades computes this coverages (computed on the asembly graph) might differ slightly to the coverage that you get from the bam file (computed on the linear sequence of the contigs)

Hey hive mind, we have just established a clean room to process archeological specimen for aDNA extraction. After intense cleaning we swapped every part of the lab as a sanity check that no DNA traces are left. Our work focuses on ancient microbes, but modern microbes have been handled before in that lab. We processed all swabs using the single-tube library prep (Carøe 2017). All libraries have as expected very low DNA concentration (≤0.2 ng/uL) and long fragments (~3kb). We would like to know if those fragments are of concern for our planned experiments. We wonder to what extent those long fragments will provide any results on a miseq run? Will a library with such long fragments provide any results? How do people infer modern contamination (long DNA fragments) in blanks? Sorry for this naive question but my wet lab aDNA xp is still at its infancy 😉 Suggestions are highly welcomed.
*Thread Reply:* You’ll have a sequencing bias towards shorter fragments (at least on Illumina MiSeq), so you will “under-sequence” the long ones. We have also attempted to use long vs short fragments in our bioinfo decontamination pipelines but since most of our samples are rather historical (only a few decades to a few hundred years old), this approach has not been too successful for us, as long genuine fragments are also present in our data. I would think that “discriminating” against long fragments might be more efficient when working with truly old samples
*Thread Reply:* Thanks Katerina. Our main concern is to what extent 3kb fragments will be sequenced at all given no shorter fragments are present? All we want to know is which organism (DNA) entered our blanks.
*Thread Reply:* Are these in your blanks as well as your libraries extracted swabs? (I.e. do you have a blank created from just the reagents, with no swab, not even a clean one). Just asking in case these very long fragments are coming from the swabs themselves (and not from the surfaces you have swabbed). I've never seen fragments of that size in blanks (although I work mostly with human DNA)
*Thread Reply:* These libraries are made from swabs used to sweep different surfaces in the newly established clean room. Only few of the libraries have fragments of that size, making it unlikely to originate from the actual swab.
We did not process any sample. Those blanks are a sanity check to make sure the clean room is clean indeed.
*Thread Reply:* My q is if we will receive reads from a miseq after loading a library with 3kb fragments.
*Thread Reply:* Oh, I see! Well, good that the swabs can be ruled out. This paper seems to suggest that 3kb amplicons from E. coli can be sequenced on a MiSeq (figure 5d), but I don't have any direct experience with this, so I'll defer to others who do and will keep my fingers crossed for you for figuring this out!

*Thread Reply:* Thx again Hannah. the figure refers to input fragments sizes for bead-bound tranpoases but not fragment sizes in the actual library used for sequencing (that is shown in 5c with most fragments < 1000b regardless of input size). in such protocols the transpoase fragments the DNA and adds seq adapter in one step. But I will dig again a bit deeper in the literature and see what I find.
*Thread Reply:* I approached the seq center and they were optimistic it will work on a miseq. apparently long fragments makes cluster localization fuzzy (leading to low base qualities), but the density on a miseq is not that high (instead of for example a NovaSeq). Will drop here what comes back.
*Thread Reply:* Just to close this topic. Seq on Miseq of our long fragments was no problem.
Hey all 🙂 I have some questions about reporting the breath and eveness of coverage. 1) Do you use a breath of coverage cut off in your work, ie. only be happy if it's over a certain percentage? 2) is there a metric you report for eveness of coverage? Right now I am visualising with samtools coverage, which does provide metrics but they are separated by accession within the genome. Thanks!
*Thread Reply:* For 1) I wouldn't do a breadth of coverage cutoff per se, but rather use it as a guide.
In some cases reference genomes will be of 'odd' pathogenic isolates that have lots of extra genes which aren't in most natural species, so by setting such a cut off could make you lose actual true positives
*Thread Reply:* So you do sort of have to look at both X coverage and % coverage in tandem, as in if you have low X coverage you ver ylikely would have low % coverage (But it's not a 1:1 ratio...)
*Thread Reply:* Hi @Ele,
First, I typically do not use a breadth of coverage (percent of bases covered at least once) cutoff because even if say ~3% of ref genome covered (which seems low), it might still be worth following this hit up because what really matters is that the few reads all come from unique places of ref genome and do not form piles of reads at a few conserved across species regions. So one might get ~3% breadth of coverage just because it was a shallow sequenced sample. The number is less important than a visual inspection.
Second, evenness of coverage (much more informative compared to breadth of coverage) is difficult to summarize in a single metric. As well as a deamination pattern. Therefore, I firmly believe, an evenness of coverage plot should be equally "a must" when reporting your findings together with a deamination plot. Currently, it is common to demonstrate deamination plots but unfortunately not coverage plots. I personally tried to create heuristics summarizing the shape of a coverage plot (I bin a ref genome and count in how many bins the coverage is equal to zero, there should be just a few such bins but not too many), but they all do not seem optimal, so I would vote for always presenting a coverage plot. For example the way @Meriam Guellil did, she shared here a link to github with her codes once
*Thread Reply:* Here you go: https://github.com/MeriamGuellil/aDNA-BAMPlotter
*Thread Reply:* Thank you all that's very helpful 😄 I'll have a go with the bam plotter 😄 Does anyone have tips for dealing with doing a similar thing for larger (non-microbial) genomes?
*Thread Reply:* @Ele working on sedaDNA samples where a goal might be to detect e.g. mammalian organisms, I typically just plot the number of reads mapping to each chromosome as a barplot. This gives some indication that there is no excess of reads mapped to a particular chromosome (unless it is the longest chromosome) but the reads should be spread more or less uniformly across the chromosomes
*Thread Reply:* That sounds like something I could do! Thanks so much Nikolay 😄

Hello, I am trying to run eager, but trying to skip adapterremoval since my samples have already been trimmed with cutadapt and merged with FLASH. The files are now ending in .all.fastq.gz. My samples were initially paird-end, double stranded, 2 colour_chemistry but now essentially single-end fastq file.
I tried running it as double end and double stranded and it gave me the following error. No TSV file provided - creating TSV from supplied directory. Reading path(s): /crex/proj/snic2021-23-584/private/Ivany/dna/03-preprocessing/cutadapt/fastq.gz
Generating paired-end FASTQ data TSV [nf-core/eager] error: Files could not be found. Do the specified FASTQ read files end in: '.fastq.gz', '.fq.gz', '.fastq', or '.fq'? Did you forget --single_end?
Should I ran eager with "single_end": true, ???
*Thread Reply:* If they are already merged, then yes 🙂
*Thread Reply:* thanks 😅

Hi everyone 🙂 What is the (minimum) # of TOTAL reads people generally target for screening with the hope of picking up pathogens/specific microbes? obv this is very context dependent, but a rough idea would be helpful. 1M reads vs 5M vs 100k
is there a minimum sequencing amount to even bother pathogen screening e.g. 100k reads / MiSeq data? any thoughts or experience would be incredible!
*Thread Reply:* I believe at EVA at least screening is either 5m or 10m? (Def. 10m for microbiome)
@aidanva @Alina Hiss @Megan Michel is it 5m still for pathogens?
*Thread Reply:* @Gunnar Neumann too
*Thread Reply:* yes, we still go for 5M if it’s from ShotGun data.
*Thread Reply:* yes shotgun. 5M is definitely more than I had envisioned so can plan for more. Excellent, thanks!
*Thread Reply:* Note thats coming from us who have cheap/routine sequencing with an internal core facility, it might be good to get input from others
*Thread Reply:* I'm going to askkk...
@Meriam Guellil @Miriam Bravo @Kelly Blevins @Åshild (Ash) @Shreya @Pooja Swali
*Thread Reply:* We're screening 5 million - I find that if it doesn't show up in the 5M (through kraken), then it tends not to even be good enough for capture (like very poor coverage/noisy damage etc) but this is just me and others may think different
*Thread Reply:* I agree for bacterial species but for viral ones it can be a bit trickier so we usually do more. Plus it's also a matter of if you are only interested in knowing what you can get full genomes of easily or what is/could be in the sample.
*Thread Reply:* Good point ☝️ I'm not looking for viral stuff
*Thread Reply:* 5M is generally a good number, although, echoing what Meriam said, if you want to screen for viruses then the more SG data you have to screen the better.
*Thread Reply:* I feel a collaborative micro blog post coming along (ping @Ele @Shreya)
*Thread Reply:* hmmm viral reads are not a priority at the moment enough to push screening higher than 5M
these perspectives were super useful, thanks everyone 🙏
*Thread Reply:* @Kevin Daly you might have volunteered yourself to write the micro blog post (basically summarise this thread ;))
*Thread Reply:* yes I love it, popping a link to this thread in the blog channel to keep tabs on it!
*Thread Reply:* Late to the party, but I sequenced my SG libs to 8-10 million just because of how many samples I had and the HiSeq lane capacity. I was able to predict libraries that performed well for capture using pretty crude competitive mapping and breadth of ref coverage measures. I was only searching for M. tuberculosis though, so haven’t run the data through a pathogen database to see what all hits. Although I’m investigating a lib that seems positive for T. pallidum that was sequenced to 10 million but only has 31 reads mapping across the T. pallidum ref, so unsure if they would have been picked up if it were sequenced any shallower. Don’t know how/if a capture will perform though. I imagine your positive flags are heavily influenced by how deeply you sequence when you have a rich background of contaminant DNA hogging your clusters. When your target pathogen is <.001%, stochasticity is bound to affects things.

Hello people 🙂 do you know if we must do or are you doing normalisation of read counts in your OTU tables as a standard procedure? if so any recommendation on how you do it? Thanks 🙂
*Thread Reply:* What sort of data and what purpose?
*Thread Reply:* Data are just kraken reports merged in a unique table so e.g. counts of reads in each species per each sample. Purpose for source tracking, metagenome composition..etc
*Thread Reply:* Is this microbiome data? Or just general pathogen screening for example?
*Thread Reply:* Source tracker you don't want to normalise
But for many other analyses (like classic stats) yes, would recommend it. @irinavelsko and I are strong proponents of CLR/ILR transformations due the CoDa principle (if you check my PNAS paper from last year I waffle on about that a bit in the SI)
*Thread Reply:* Pathogen screening I wouldn't normalise either
*Thread Reply:* It doesn't help for anything really (that I can think of anyway?
*Thread Reply:* Ok I see it makes sense for the stats 🙂 thanks! I found that section in your paper 🙂 I will have a look!
*Thread Reply:* @Maria Lopopolo and @James Fellows Yates CLR / ILR transform is used on compositional data, i.e. when you for example normalize your count matrix by library sizes, then your counts per library sum up to a certain number (like 1), so this is a compositional data, then indeed you should run a CLR / ILR on the top if you want to proceed with traditional stats like PCA, differential abundance, linear model etc. But Kraken counts that @Maria Lopopolo mentioned are not compositional data unless you normalize the raw counts by library sizes. In contrast, QIIME or MetaPhlan by default give you an abundance / OTU which is compositional, i.e. already normalized by library sizes, so-called TSS - total sum scaling normalization
*Thread Reply:* Basically, my recommendation would be to normalize raw Kraken counts by TSS (library sizes) or CSS (more intelligent, cumulative sun scaling) and preferably by genome length (if you also want to compare abundances across species). Then you need to do CLR / ILR only on the top of TSS counts (compositional data), and can skip it at all if you use CSS normalization or genome length normalization (with either TSS or CSS). Sorry for very complicated explanation 😞
*Thread Reply:* Thanks @Nikolay Oskolkov does this apply to krakenuniq too?
*Thread Reply:* Yes! Short summary: some library size normalization should be used, then you data may or may not become compositional (when abundances per library sum up to 1 for all libraries). If your abundance is compositional, then use CLR / ILR on the top, otherwise skip it
*Thread Reply:* Hm, if I'm following correctly, but raw read counts are compositional, as you're fixed by the capacity of the sequencing lane (which is a 'synthetic' cap)? I thought the CLR transformation is basically converting the variablty in the totals across different seq depth samples to make them compatible, i.e. I thought the CLR was doing the normalisation for you?
*Thread Reply:* @James Fellows Yates as far as I understand it, CLR is just a trick to transform your data from simplex space (compositional data) to euclidean space. Otherwise you e.g. have to run a PCA with aitchison (and not euclidean) distance. I agree that in theory many types of sequencing data are compositional, however as long as counts per library / sample do not strictly sum up to 1 across all libraries / samples, you should not worry about CLR. Otherwise, CLR should also be used e.g. in RNAseq, but I do not think people in this field heard about CLR 🙂
*Thread Reply:* Hmm ok... Im too tired to discuss it further now, but from what I remember and what I checked in these two reviews, they seem to imply you just need to give raw counts, each sample does not need to sum up to one:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6755255/ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5695134/#!po=6.94444|https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5695134/#!po=6.94444
But it would be cool to talk about this in the future, as I imagine you understand this stuff just better than I do (I suck at maths and stats)
RNASeq people do know about CLR stuff but at that point they had equal or better performing methods such as DESeq
*Thread Reply:* (sorry to hijack the thread @Maria Lopopolo)
*Thread Reply:* I also remember TSS being an alternative to CLR not applying it on top of (as also implied here: https://www.frontiersin.org/articles/10.3389/fams.2022.884810/full)

*Thread Reply:* No worries @James Fellows Yates and @Nikolay Oskolkov these are the conversations that probably will be useful to many and that I need to understand too (even if you are speaking chinese rn). I guess one more topic to discuss for common mortals in SPAAMtisch :mask_parrot:
*Thread Reply:* Note I'm generally agreeing with @Nikolay Oskolkov otherwise ;)
*Thread Reply:* Just whether CLR/ILR is an alternative or on top of TSS for differential abundance of raw counts.
Otherwise like he said MetaPhlAn has their own normalisation
*Thread Reply:* Thanks @James Fellows Yates, I certainly do not have a good literature overview in this field, and therefore appreciate to know your point of view. However, I have my own experience of trying to understand why compositional data are generally considered to be “dangerous” in mathematical statistics. It has something to do with features (microbes, genes etc.) becoming artificially correlated for compositional data. In this post I tried to demonstrate how this “spurious” correlation (discovered by Karl Pearson) can arise when a random count matrix is library size (TSS) normalized: https://towardsdatascience.com/pitfalls-of-data-normalization-bf05d65f1f4c (sorry for self-promotion). It is a super-simple code that everyone can reproduce and see how uncorrelated features become “spuriously” correlated after TSS. Basically TSS should never be used because of that reason, but it is still widely used in metagenomics / 16S. Therefore to fix it people came up with the CLR-transform which is (if you check) a combination of log-functions and thus resembles the aitchison distance, which is a more proper distance metric when a constraint has been applied to a data (and summing up counts to 1 is a constraint). In RNAseq people used library size normalization in the early days but quickly gave up because of the danger of compositional data, and in addition equalizing sample / cell libraries seemed like an over-normalization that destroyed all the interesting heterogeneity across samples / cells


*Thread Reply:* @Nikolay Oskolkov I am bit confused on how library size normalization would not make your microbiome compositional. Based on my understanding, I agree with James that sequencing capacity (along with other non-biological factors) will always make your data compositional. For instance, we can never 'know' whether the zeros in our datasets are due to sequencing capabilities or because they are biologically not present there for a reason. Could you clarify or elaborate more on how the data is not compositional.
*Thread Reply:* Applying CLR on raw counts looks good to me too. It seems similar to using a simple log-transform on raw counts that makes count data more “euclidean”. In scRNAseq for example this simple log-transform is considered as a weak normalization that brings counts from different libraries on the same scale which is often sufficient. Although I do not think that log-transform actually properly equalizes library sizes, I believe it is better than nothing
*Thread Reply:* @Sterling Wright compositional data is data where rowSums or colSums (in R language) results in the same fixed value (often 1) for all raws / columns. You can take Kraken counts from multiple samples and check whether summing up counts (per sample) across all microbes results in the same fixed value for all samples. I bet it will not. So your Kraken data are not compositional (at least in mathematical sense). Now you can take MetaPhlan (default settings) abundances and run rowSums / colSums, I bet you will get same fixed value for each of your samples. So your MetaPhlan data are compositional and you should not naively run PCA on compositional data. Now, here comes the question: both Kraken and MetaPhlan were run on the same fastq-files. How come the same data become compositional for MetaPhlan and do not for Kaken? The thing is that, MetaPhlan was normalizing the raw abundance counts with library sizes (without telling you), while Kraken did not do it. So irrespective of sequencing capacity constraint, one tool gives you compositional and the other one gives non-compositional data. Again, I am talking about mathematical definition, not the biological natural constraints
*Thread Reply:* Library size normalization will always give you compositional data while sequencing (lane capacity) does not result per se in compositional data (at least the raw counts do not sum up to one unless they have been bioinformatically library size normalized) 🙂
*Thread Reply:* Okay I see how that would make mathematical sense. I have to admit that I have not dealt with Kraken datasets, only MALT. And so this maybe one of the reasons on why I am confused. However, out of curiosity, could you explain whether this library size normalization procedure would be impacted if you changed the sequencing depth on the same dataset. I have a clearer understanding for this the log transformation procedures. CLR/ILR/ALR transformations are applied to feature/OTU tables because if you stuck with count data you are going to get spurious correlations as soon as you change the sequencing depth. If you were to remove one read or count of a feature, then the proportion of all other features go up and the proportion of that one feature decreases. On the surface, you would think that all the other features are positively correlated with each other even though their counts did not change and only the selected feature did.
*Thread Reply:* Very nice Thread here! And very relevant and important for SPAAM
*Thread Reply:* I am totally with @Nikolay Oskolkov in all that he explained. This is what I have always done when analyzing metagenomic data (i.e., some sort of normalization by sequencing effort per sample, to make data compositional) and then some sort of transformation
*Thread Reply:* the simplest I was doing was to normalize by the total number of reads per sample and then using the VST transformation in DESseq2
*Thread Reply:* but I think that what Nikolay proposed is probably better
*Thread Reply:* in any case, in my opinion comparing datasets with very different sequencing depths is always risky, because of the 0s that Sterling was mentioning. You are not able to see the same richness in highly and lowly sequenced samples
*Thread Reply:* so some considerations on the taxa to be measured should be considered.
*Thread Reply:* Some people downsample reads in samples to the same number of read counts per dataset (QIIME was doing that when I used to use it)
*Thread Reply:* @Sterling Wright I believe you see your abundance data as proportions / fractions, not absolute counts. This is similar to 16S / OTU / QIIME terminology. If your abundances are fractions then they naturally sum up to 1 and the microbes are correlated by that constraint. But I think the constraint that resulted in the proportions is a computational thing, i.e. if one wouldn’t have applied library size normalization, the data would not have been strictly compositional
*Thread Reply:* I think that it would be great that the SPAAM community converge to a recommended practice around this topic, for instance, to compare ancient microbiome samples for beta and alpha diversity, to identify taxa that are significantly changing between conditions, etc.
*Thread Reply:* If the input data is not properly treated, then results are just meaningless
*Thread Reply:* I agree very much. If we want our results to be reproducible and try to minimize the chance of results being inconsistent due to pipeline differences, I think there is a lot of work that needs to be done in this space.
*Thread Reply:* @Nikolay Oskolkov I'm catching up now with more sleep - so technically you're saying if you do TSS scaling you must use something like CLR.
However, I guess my question is must you do TSS scaling to do CLR, or can you apply CLR directrly on raw counts (which you appear to imply above, and is what most of the tutorials/microbiome papers on the topic also seem to suggest)?
also don't feel shy to self-promote, your blog posts are very good
*Thread Reply:* Hi @James Fellows Yates, apologies for the delay, I am also trying to find time to sleep (sometimes) with my three kids 🙂 I agree, you must always use CLR after TSS, and you must not do TSS in order to be able to run CLR 🙂 However, a simple CLR alone does not remove the dependence of microbial abundance on sequencing depth, therefore you should equalize somehow library sizes via TSS or CSS to correct for the technical variation in sequencing depth between your samples.
I took a Kraken abundance matrix from one of my project and compared how a mean abundance count changes vs sequencing depth (it should not depend on sequencing depth) for unnormalized, CLR normalized, library size normalized (TSS), and library size+CLR normalized counts, please see below. I am sorry, I can not share the abundance matrix, but I am sharing the code I used below so that you could understand how the plots were computed, and you could also check how the plots look like for a Kraken abundance matrix (columns are samples and rows are microbes) from your favorite project.
```library("compositions")
Reading, adding offset +1 (to get rid of zeroes), displaying and storing library size information
df<-read.delim("abundancematrix.txt",header=TRUE,row.names=1,check.names=FALSE,sep="\t") df<-df+1 head(df) libsize<-colSums(df)
par(mfrow=c(2,2))
Unnormalized data
plot(log10(colMeans(df)+1)~log10(libsize+1),main="Unnormalized data",ylab="log10 ( Mean Count )",xlab="log10 ( Sequencing Depth )") abline(lm(log10(colMeans(df)+1)~log10(libsize+1)))
CLR-transformed data
dfclr<-clr(df)+20 head(dfclr) plot(log10(colMeans(dfclr)+1)~log10(libsize+1),main="CLR normalized data",ylab="log10 ( Mean Count )",xlab="log10 ( Sequencing Depth )", ylim=c(1.25,1.45)) abline(lm(log10(colMeans(dfclr)+1)~log10(libsize+1)))
Library-size normalized data
dflibsize<-matrix(nrow=dim(df)[1],ncol=dim(df)[2]) for(i in 1:dim(df)[2]) { dflibsize[,i]<-(df[,i]/sum(df[,i])) } head(dflibsize) plot(log10(colMeans(dflibsize)+1)~log10(libsize+1),main="Library size normalized data", ylab="log10 ( Mean Count )",xlab="log10 ( Sequencing Depth )") abline(lm(log10(colMeans(dflibsize)+1)~log10(libsize+1)))
Library-size normalized + CLR-transformed data
dflibsize<-dflibsize+1 dflibsizeclr<-clr(dflibsize)+20 head(dflibsizeclr) plot(log10(colMeans(dflibsizeclr)+1)~log10(libsize+1),main="Library size + CLR normalized data", ylab="log10 ( Mean Count )",xlab="log10 ( Sequencing Depth )",ylim=c(1.25,1.45)) abline(lm(log10(colMeans(dflibsizeclr)+1)~log10(libsize+1)))```

*Thread Reply:* From the figure above, my conclusion is that, CLR alone is not sufficient to correct for technical variation in sequencing depth
*Thread Reply:* Thanks @Nikolay Oskolkov (alwasy prioritise sleep over SPAAM btw ;)), but those plots are really helpful to demonstrate. This would be another nice blog post I think.
So to conclude for @Maria Lopopolo (Nikolay correct me if I'm wrong) -
Pathogen screening - no normalisation needed
Microbiome work: normalisation already happens for some tools for you (MetaPhlAn3, etc). But if you have raw read counts (Kraken2/MALT etc.) it is possible for a quick and dirty normalisation to just run a CLR/PhILR transformation to do basic exploratory analyses (maybe PCA, for example). However to do it properly, i.e. before doing full statistical tests for differential abundance to make biological inference from, you should normalise by TSS/CSS then perform a log ratio transform (CLR/PhILR) etc.
*Thread Reply:* Yes, good summary @James Fellows Yates!
*Thread Reply:* (now we wait for @Shreya and @Ele to bug us 😉 but maybe we could do a little collab. one between the three of us?),
*Thread Reply:* @James Fellows Yates and @Nikolay Oskolkov thank you so much for this! I appreciate you both taking the time to disentangle this from my no-stupid-question 😊
*Thread Reply:* Clearly not a stupid question!
*Thread Reply:* It was an awesome thread! Very thanks to @Nikolay Oskolkov!
*Thread Reply:* Hi everyone thanks for the fantastic thread! (I managed to read through only now). We also took as routine to do TCC+CLR for microbiome data. Quick question @Nikolay Oskolkov (and the others), so after clr-normalization which distance can be used (e.g for cluster analysis)? Bray-Curtis? Or better go for Aitchinson distance on the non-clr-normalized data? (is it eventually equivalent? I never really tested that systematically but it seems not).
*Thread Reply:* @Claudio Ottoni I personally have not tested Bray-Curtis (on TSS+CLR normalized) vs Aitchison on TSS counts for clustering, so do not unfortunately have a good answer 😞 Many things to test but very little time...
*Thread Reply:* @James Fellows Yates and @Nikolay Oskolkov going back to this much discussed topic eheheh what to do when your geometric mean it’s 0 bc many species in the table have “0” for many samples? Is it here that you consider filtering out species that are 0 for e.g > 50% of the samples?
*Thread Reply:* @Maria Lopopolo geometric mean on zero-inflated matrix of counts is problematic to compute (as you said it easily becomes zero if at least one element in the vector is zero). Therefore, for example, people do not use DESeq / TMM normalization (which are based on computing geometric means) for single cell RNAseq (zero-inflated) while DESeq / TMM are gold standard for bulk RNAseq (non-zero-inflated). Yes, for zero-inflated data (and I would say metegenomic data are zero-inflated) I would either harshly filter lowly abundant species (to justify the use of geometric mean normalization) or use some normalization that is not based on geometric means

Hi people:) how much it would cost for per sample from extraction to sequencing in an aDNA lab in your case? I was late to know and start a grant application due on tomorrow night, and I need to quote the spending, so it would be great if you can share the info with me. Thanks!!
*Thread Reply:* Hi Yuti, Could you provide a little more information? Do you plan on using shotgun sequencing or capture? What type of samples will you be looking at?
*Thread Reply:* Hi Sterling, I plan to use shotgun, and it’s dental calculus
*Thread Reply:* Got it. We budget $20 per sample for dna extraction and $120 per sample for double stranded libraries. But a big question will be how deep you want to sequence your samples and how many samples you have. Sequencing deeper depths will be better but will be more expensive. Do you have an idea which sequencer you will use? Also do you have an idea about the preservation of the samples
*Thread Reply:* Thanks for the info Sterling!! I have 29 samples, and to be honest I have no idea yet how deep I am going to sequence, I only worked with live host samples before, the dental calculus samples I have now are from primate collection in museum. I think I will say around 100~150 for the grant purpose so far
*Thread Reply:* Got it. With that many samples and that much money, you should have enough for a hiseq run, maybe even a novaseq run. This does depend on where you send your samples and what kind of contracts your institution has with sequencing facilities. But hopefully this helps
*Thread Reply:* I second these estimates! We budget $150 for extractions plus double stranded libraries per samples and then the major cost is sequencing, which will vary based on depth of coverage desired and prices at the sequencing center you work with

Hi all! Maybe I've just missed this in the documentation, but for users of the new aMeta pipeline, which databases are required and which are optional? Followup simple unix question - Is there an efficient way to "wget"/download these via command line? I'd prefer not to download GBs of data locally just to move them to a remote cluster later. Thanks!
*Thread Reply:* Scratch part 2 - figured it out with a clever renaming on command line!
*Thread Reply:* @Zoé Pochon @Nora Bergfeldt @Nikolay Oskolkov 👆
*Thread Reply:* Hi @Carly Scott, sorry for the delayed reply! If you follow a simple installation via:
1) git clone <https://github.com/NBISweden/aMeta>
2) cd aMeta
3) mamba env create -f workflow/envs/environment.yaml
4) conda activate aMeta (alternatively if this fails, try conda activate ancient_microbiome_workflow)
5) cd .test
6) ./runtest.sh -j 4
there is a toy KrakenUniq database and Bowtie index built for testing purposes. Please make sure that everything works for you on the testing step. For real world projects you should use one of the following databases here:
KrakenUniq database based on full NCBI NT: <https://doi.org/10.17044/scilifelab.20205504>
KrakenUniq database based on microbial part of NCBI NT: <https://doi.org/10.17044/scilifelab.20518251>
KrakenUniq database based on microbial part of NCBI RefSeq: <https://doi.org/10.17044/scilifelab.21299541>
Bowtie2 index for full NCBI NT database: <https://doi.org/10.17044/scilifelab.21070063>
Bowtie2 index for pathogenic microbial species of NCBI NT: <https://doi.org/10.17044/scilifelab.21185887>
I personally would recommend to use the microbial NCBI NT (if you are after microbes only and less after eukaryotes) or full NCBI NT (if you are after both eukaryotes and prokaryotes) for KrakenUniq part of the workflow, and full NCBI NT Bowtie2 index for following up the kmer classification. You should not be worried about the MALT followup step as this should be tuned automatically, i.e. the database is built dynamically for each project. If you run into installation / execution or resource issues, please let me know
*Thread Reply:* Awesome, thanks for the info! This clears thing up - I'll let you know if I bump into anything else as I run it 🙂
*Thread Reply:* Quick followup: What is "PathogensFound.tab"? Is this a list of potential pathogens that should be fed into Kraken or the output of Krakenuniq?
*Thread Reply:* MissingInputException in rule Filter_KrakenUniq_Output in line 25 of /scratch/06909/cbscott/ancient_metagenomics/aMeta/workflow/rules/krakenuniq.smk:
Missing input files for rule Filter_KrakenUniq_Output:
output: results/KRAKENUNIQ/S30455/krakenuniq.output.filtered, results/KRAKENUNIQ/S30455/krakenuniq.output.pathogens, results/KRAKENUNIQ/S30455/taxID.pathogens
wildcards: sample=S30455
affected files:
resources/pathogensFound.tab
*Thread Reply:* ^is what I'm specifically bumping into - I'm just not sure what input could be missing here, if you have any insight?
*Thread Reply:* Hi @Carly Scott, yes, this is our very permissive custom list of microbial pathogens that was built based on extensive literature search. This file should be present at the aMeta github repo, I will double check this, could you please also locate this file in your cloned repo?
*Thread Reply:* Thanks, got it @Nikolay Oskolkov - it is subset in the hidden ".test/resources" folder.
*Thread Reply:* Though this looks like a small subset of potential species (just the test file). Should these files have been downloaded through the configuration of the software? In a fresh clone of the git repo in my environment I'm not seeing it.
*Thread Reply:* @Carly Scott I have checked this and this file is apparently not in the aMeta repo but it is in the PathoGenome https://doi.org/10.17044/scilifelab.21185887 that we published together with the workflow. You can download the pathogensFound.very_inclusive.tab file, put it to aMeta/resources and provide the path to the file in the config.yaml via pathogenomesFound: resources/pathogenomesFound.tab
*Thread Reply:* sorry about it, we should have included this file in the resources-directory in the aMeta repo, I will open an issue for that
*Thread Reply:* How the config file in a project folder looks like: ```samplesheet: resources/samples.tsv
analyses: mapdamage: true authentication: true malt: true
Databases !!!! CHOOSE BETWEEN FULL_NT AND MICROBIAL
krakenuniqdb: /proj/nobackup/metagenomics/databases/DBDIRKrakenUniqMicrobialNTPlus_CompleteGenomes
krakenuniqdb: /proj/nobackup/metagenomics/databases/DBDIRKrakenUniqFullNT
bowtie2pathodb: /proj/nobackup/metagenomics/databases/PathoGenome/library.pathogen.fna pathogenomesFound: /proj/nobackup/metagenomics/databases/PathoGenome/pathogensFound.veryinclusive.tab pathogenomeseqid2taxiddb: /proj/nobackup/metagenomics/databases/PathoGenome/seqid2taxid.pathogen.map maltseqid2taxiddb: /proj/nobackup/metagenomics/databases/DBDIRKrakenUniqFullNT/seqid2taxid.map.orig maltntfasta: /proj/nobackup/metagenomics/databases/DBDIRKrakenUniqFullNT/library/nt/library.fna maltaccession2taxid: /proj/nobackup/metagenomics/databases/DBDIRKrakenUniqFullNT/taxonomy/nuclgb.accession2taxid ncbidb: resources/ncbi kronadb: /pfs/data/databases/KronaTools/20220207/
Additional config
nuniquekmers: 1000 ntaxreads: 200```
*Thread Reply:* Yes, thank you @Zoé Pochon! @Carly Scott needs to change this line "pathogenomesFound: /proj/nobackup/metagenomics/databases/PathoGenome/pathogensFound.very_inclusive.tab" after she has downloaded pathogensFound.very_inclusive.tab file from https://doi.org/10.17044/scilifelab.21185887
*Thread Reply:* Thank you @Nikolay Oskolkov and @Zoé Pochon - I missed it by downloading the full nt bowtie index instead of the microbial file. I've got it now - I'll let you know if it works 🙂
*Thread Reply:* Hi @Carly Scott, we updated the docs (README) for aMeta https://github.com/NBISweden/aMeta, it should now be much easier to install and quick start running it on your samples. We put references for all databases mentioned in the configuration file, so hopefully you can download them and run aMeta. Please let me know if you still experience difficulties and I will assist you
*Thread Reply:* Thanks for the update @Nikolay Oskolkov. So far things seem to be running on my end - I appreciate the guidance. A quick followup, as I'm working with non-human samples: is the literature-search for PathogensFound based on pathogenic microbes specifically in humans or across taxa?
*Thread Reply:* Glad it is working for you @Carly Scott! The pathogenic microbes are not human-specific, many of them are plant and animal pathogens. The list is very comprehensive and permissive in order not to miss anything interesting, however some manual (common sense) verification is still required. So the fact that they are called "pathogens" in our list does not fully guarantee that they really are, but they rather should be considered as good pathogenic candidates. Do not hesitate to contact me if you have any question or problems with aMeta 🙂
*Thread Reply:* Hi again! Is there a way to turn adapter trimming off for aMeta - I've already trimmed my files and removed human contamination, which I think is causing the following error:
*Thread Reply:* Job 2: FastQC_BeforeTrimming: RUNNING QUALITY CONTROL WITH FASTQC FOR SAMPLE /work/06909/cbscott/ls6/aDNA_2022/S30455.short.fastq BEFORE TRIMMING ADAPTERS
Reason: Missing output files: results/FASTQC_BEFORE_TRIMMING/S30455_fastqc.zip
Waiting at most 5 seconds for missing files.
MissingOutputException in rule FastQC_BeforeTrimming in line 1 of /scratch/06909/cbscott/aMeta/workflow/rules/qc.smk:
Job Missing files after 5 seconds.
*Thread Reply:* Hi @Carly Scott could you post me a few lines from the samples .tsv file? It does not seem like an error due to adapter trimming because this is a QC before adapter trimming, i.e. the very first step of the workflow. I believe the workflow can not find the input files, therefore would like to see your samples.tsv
*Thread Reply:* Ah! I've got it. You're right it wasn't a trimming issue, it's a file naming convention issue. Sorry to bug yoU!
*Thread Reply:* I named my files NAME.descriptor.descriptor.fastq, and it seems they didn't parse correctly
*Thread Reply:* Adapter trimming does not take much time and from my experience trimming adapters once might result in some adapter left overs, so trimming an extra time can be only beneficial
*Thread Reply:* No problems at all, do not hesitate to post your questions here or you can send me a private message as well
*Thread Reply:* Hi @Nikolay Oskolkov, apologies to bother you again! I am trying to run aMeta with a custom database (of marine microbes), and have built my botwtie2 database separately. However, aMeta insists on overwriting these files and recreating the bowtie2 database again, which is really slowing down the software. Do you have any insight to why it might be doing this? I'm fairly certain the database is complete, despite the error message, as the job completes without any issues on my end (using the same script as aMeta, but parallelized).
*Thread Reply:* bowtie2-build-l resources/bt2db/full.library.fna resources/bt2db/full.library.fna
*Thread Reply:* The overwrite process:
[Thu Dec 1 17:45:31 2022]
rule Bowtie2_Index:
input: resources/bt2db/full.library.fna
output: resources/bt2db/full.library.fna.1.bt2l, resources/bt2db/full.library.fna.2.bt2l, resources/bt2db/full.library.fna.3.bt2l, resources/bt2db/full.library.fna.4.bt2l, resources/bt2db/full.library.fna.rev.1.bt2l, resources/bt2db/full.library.fna.rev.2.bt2l these outputs already exist in the directory, from my local build
log: logs/BOWTIE2_BUILD/resources/bt2db/full.library.fna.log
jobid: 6
reason: Forced execution
resources: tmpdir=/tmp
*Thread Reply:* Hi @Carly Scott, thanks for reporting, please do bother me with your questions, we want to improve aMeta! Regarding your question, did you use --forceall flag? If so, snakemake will try to execute absolutely all rules even if some of them can be skipped like for the case of pre-built custom Bowtie2 index
*Thread Reply:* Thanks!
I did not use the --forceall flag. I ran it with: snakemake --snakefile workflow/Snakefile -j 10
*Thread Reply:* @Carly Scott it looks like your resources/bt2db/full.library.fna file is newer than the index (**.bt2l files), can it be the case? Only in this case aMeta tries to rebuild the index. I am also puzzled about the reason: Forced execution , this message means that you must have used --forceall, but if you did not, this sounds strange
*Thread Reply:* @Carly Scott would you mind checking the creation dates of your resources/bt2db/full.library.fna and the index (**.bt2l files) in the resources/bt2db folder?
*Thread Reply:* Ah you know what might have happened - I created all of the files in a secondary location to keep them from being overwritten, and then created a copy in resources/bt2db (cp my_backup_dir/full.library.** resources/bt2db), which put the same timestamp on all of the files (see screenshot). Let me redo this by moving first full.library.fna over, followed by the .bt2l files and try rerunning.



*Thread Reply:* I'll create the bt2 index in the directory and see what happens. I'm also not opposed to letting aMeta recreate it, but it's not a memory intensive task for this database, so I've been parallelizing it separately from aMeta
*Thread Reply:* Please try to use the --rerun-incomplete flag and see whether aMeta still insists on rebuilding the index
*Thread Reply:* iT does - this is what I started with during the troubleshooting, but I can send over the message if you would like.
*Thread Reply:* @Carly Scott your custom Bowtie2 database does probably not match the KrakenUniq DB? I mean, those two had presumably different reference fasta files, or did you build a KrakenUniq DB from the same resources/bt2db/full.library.fna?
*Thread Reply:* So full.library.fna is a composite of ~1,000 genomes which I catted together. I think I built the KrakenUniqDB based on the individual genomes in a directory, but the bowtieDB on the full.library.fna. I can go back and build them based on the same reference
*Thread Reply:* Generally does aMeta need identical Kraken and Bowtie databases?
*Thread Reply:* @Carly Scott yes, KrakenUniq, Bowtie2 and Malt databases should use exactly the same fasta-file (same headers and seqs). By design, aMeta was not a collection of tools that can be used individually, but they all are supposed to work together and be consistent with each other. Therefore, it is not straightforward to replace a Bowtie2 index without modifying the KrakenUniq DB, unfortunately. In your case, if the concatenated fasta contains exactly the same reference sequences as in the directory with the individual genomes (same headers and seqs), it seems your KrakenUniq DB matches your Bowtie 2 index, so you should not have KrakenUniq vs Bowtie2 incompatibility problems
*Thread Reply:* This all however does not seem to explain why aMeta insists on rebuilding the Bowtie2 index, let me think a bit
*Thread Reply:* Of course the easiest would be to just let aMeta to rebuild the Bowtie2 index but we need to figures out why aMeta wants it 🙂
*Thread Reply:* I'll set up a run this weekend to let aMeta rebuild the index, and let you know what happens. I can then check for discrepancies between the directory/files where I built it separately and aMeta's directory
*Thread Reply:* Good, thanks! I believe, if you built a KrakenUniq DB using a number of reference fasta-files in a directory, and then just concatenated those reference fasta-files into a single grand fasta-file and built a Bowtie2 index on it, those two DBs are compatible and you should not get troubles due to different seqIDs
*Thread Reply:* Ran just fine over the weekend letting aMeta rebuild the database. As far as I can tell the files are identical to what was there before, but the software no longer needs to rebuild the bowtie2 index at each start up. I'm just going to call this resolved on my end rather than looking into it any further for now.
*Thread Reply:* Thanks @Carly Scott, I am glad that it worked fine eventually 🙂 but we (developers) still need to fix this issue. I shared what you told me with my colleagues and we seem to have another user experiencing the same issue, so we will perhaps restrict this rule severely so that it is not activated at all if a pre-built Bowtie2 index is provided. We are currently discussing possible solutions. Thanks a lot for using aMeta and reporting this issue. Please do not hesitate to post here any other issues that you might experience in the future
*Thread Reply:* Super simple question - where should I find the final result from aMeta (a table containing microbes per sample sorted by presence + origin)? I'm trying to figure out if I have a software error or am just looking in the wrong place. The abundance tables given in "MALTABUNDANCEMATRIX_**" are pre-authentication for damage, etc., yes?
*Thread Reply:* Hi @Carly Scott, sorry for the delay! We have recently added a heatmap overview of ancient microbes, please check the “Main results” section here https://github.com/NBISweden/aMeta
*Thread Reply:* You have in general 3 abundance matrices delivered by aMeta: KrakenUniq abundance matrix, and 2 Malt abundance matrices (computed from rma6 and sam-alignments). All 3 of them do not tell you anything about authentication, therefore we made the heatmap overview (above) that demonstrates “authentication scores” (quantification of 7 quality metrics) on the top of detected microbes
*Thread Reply:* So if you only want to see what microbes are present in your samples, I would take the KrakenUniq abundance matrix. However, if you want authentication information, you will either have to manually check authentication pdf-figures (per sample per microbe) in results/AUTHENTICATION, or run our script for aggregating authentication scores on a heatmap
*Thread Reply:* @Nikolay Oskolkov Perfect, thank you! If you're interested, in the meantime I wrote a little shell script that will take the highest ranking microbes for each sample and combine all the pdf's together into one master document you can visually check locally (for lots of samples it's tedious to download them off the server separately). Then, after manual inspection I subset the abundance matrices for my "verified" sample set. Running aMeta with a custom database, I decided it would take too long to inspect all of the pdfs returned (as I've expanded beyond bacteria to other microbes which may be in my samples).
*Thread Reply:* @Carly Scott glad to hear you reached the pdf-output step, meaning aMeta seems to have worked correctly 🙂 Indeed, collecting, downloading and inspecting all the numerous pdfs is tedious, therefore we tried to automate it, but have only recently implemented the summarizing rule. I believe when you started running your samples, the summary heatmap was not yet available, so you had to aggregate the pds manually. Hopefully, in the future runs the aggregation will be much easier 🙂
@channel based on @Yuti Gao’s question above, how useful/interesting do you think an (anonymous) survey of extraction-sequencing costs across the whole field would be ?
*Thread Reply:* Blooooog.
But shotgun not doing it (free idea for someone!)

Hi all! I'm a newcomer here and, in the spirit of the channel, I'm writing to ask some clumsy, far too general questions that may betray even more ignorance than I'm already anticipating. I've reached out to a few researchers in the ancient metagenomics community and was encouraged to ask a broader audience their thoughts on a few of them. Recently, I've finished up a postdoc where I mostly performed bioinformatic analyses on metagenomic datasets from modern environmental samples, with a little HGT twist given that HGT took centre stage during my PhD in microbial evolution. I liked my work and I got the chance to collaborate on some very diverse topics, but I've always been fascinated by history and archaeology, and believe that ancient DNA work may be a good fit for me. I've been reading up on general methods, challenges in the field, and of course, exciting findings in primary research articles, but I'm worried about approaching potential PIs and being dismissed as an unserious candidate. Has anyone had any experience approaching PIs as someone from outside the field? Relatedly, how did you find your research topics? Did you come in with some very concrete ideas or did you select from projects that already had some momentum behind them? I'm very open to a range of topics but I'm concerned this may simply read as naivety about the field and work against me. Finally, does anyone have any experience with remote (or semi-remote, with periodic travelling) work and the general perception of it among PIs? Though I did wet-lab work for my PhD, my most important skills really are in bioinformatics, and anyway, the idea of being responsible for DNA extraction of some precious limited sample terrifies me (for now--never say never!). And I think we've all demonstrated an ability to work effectively from home in the last few years. The alternative is waiting a couple of years to start, since my partner is doing his postdoc currently, but I'd much rather start sooner rather than later. Any thoughts on these or anything else along this line are very welcome!
*Thread Reply:* I can't say much about approaching people directly as I've not had to od thi sfo far, but some general thoughts from me:
Outside the field is rarely an issue, it's a very small field - very few people 'grow up' in the discipline. Furthermore, often people with modern biology backgrounds are welcome, as many students come from a social sciences background
If you have your own ideas/projects, you need to bring your own funding for them, otherwise you will have to apply for existing projects. It's rare that our discipline have money 'hanging around' for 'cool ideas' (so to say)
Unless you go to larger labs (which there are relatively few often), often you will have to do everything from A-Z at PhD level anyway. I'm not sure how this is at postdoc; that said the lrager labs are more and more moving into purely analysis (as they have a lab core). I think remote working will always be a lab culture thing though rather than specific to a field
*Thread Reply:* Hi @Laura Woods, while James gave you very good field-specific comments, I can briefly share my general impression of migrating across fields as I did my PhD in theoretical physics and switched to biomedicine which was quite shocking experience 🙂 I have been viewed everyday by many as an unserious candidate 🙂 which is very frustrating, but with time I learnt to overcome this physiological barrier, your different from others background can be your strength. My experience is that the vast majority of PIs prefer talking to somebody who understands their language (terminology), but from time to time I see other PIs who are open for exploring new methods in more risky projects. In your particular case, I would not be worried much because bioinformatic analysis of modern environmental metagenomic samples is super close to what many people in this channel are doing, and you are not an outsider (in contrast to me 🙂 ). Regarding finding your research topics, during my carrier I mostly had to adjust to already established projects, so even if I have my own ideas, they are not straightforward to implement or to be supported (as James very well pointed out it has something to do with own funding 🙂 ). However, my major transition from physics to biomedicine happened when I brought my naive ideas to a group of medical doctors who for some reason found them interesting, but this was a luck and an exception. Generally, I would say, to implement own ideas one needs own funding and a secured position. Otherwise one needs to adjust and find a niche. Most of the time I am learning what other people are doing before suggesting something original 🙂 In summary, I would really encourage you to try. Motivation and interest, in my eyes, are more important than knowledge in a particular area, everything can be learnt providing you have interest and passion
*Thread Reply:* Hi @Laura Woods, I would very much agree with what has been said so far. From the perspective of a group leader in a very small group without many resources, I'd say: • having people with experience in environmental metagenomics is amazing! The ancient metagenomics field is very much a conglomerate of people coming from the aDNA branch, the microbiology branch and the bioinfo branch. Then we learn the respective other parts to make sense of our data • Approaching people will never hurt, as long as you are fine with getting (hopefully) polite replies stating that in most cases there is no money to employ a person ad hoc. • This is where bringing in own money would be important. If you identify a research group that does what you like, do reach out to them with the prospect to develop a research topic for which you can write up a scholarship application (if they don't have money available). There is a whole set of postdoc grants that would be suitable, from Marie Curie, to EMBO, HFSP, to country-specific ones. Having your own funding, even on a co-developed project, will give you a lot of independence. • In my lab, we tend to do everything from sampling to analyses, but I agree that the bioinformatic analytical expertise is usually the bottleneck. • Working remotely will very much depend on a situation, the source of funding and the lab culture. It is possible but I would think that most labs would prefer having the group members physically present. It's not only about doing the work but about having a sense of belonging together, the possibility to chat with each other about science and life. You will never have unexpected spontaneous meetings online...
*Thread Reply:* Thanks a lot for such thoughtful responses, they have been really encouraging! Actually, part of the reason that I even worked up the nerve to write this message was because I suspected that the field was generally more welcoming to outsiders, and it seems that even those with a much greater change in academic focus than mine can find success! As for finding a research project, the idea of coming up with an idea while being so inexperienced is a bit daunting anyway and I certainly haven't developed the kind nose for what projects will have broad interest or get funded properly that one develops after being in a field for long enough. Therefore, co-developing a project and applying for funding or applying for positions advertising funding are very acceptable options. Regarding remote work, it is always a big ask and to be honest, I do prefer coming into an office--I began and ended my last position in lockdown and had a constant sensation of being a bit adrift. I completely understand that group leaders would share that view. I suppose I've just come up against the old two-body problem: my partner is already working on his postdoc and we'd been a part while I did my last one, so I was hoping we could live together this time round. Ah well, no harm in asking and I can always wait until after he's finished if need be. Thanks again to all of you, I really appreciate your taking the time to answer my questions!
*Thread Reply:* Totally feel you @Laura Woods, particularly about the 2-b problem. My husband and I fought on this front for years... Happy to chat more about this and any other aspects of changing fields. My moves have not been as drastic as @Nikolay Oskolkov’s but I have been financing my biggest transitions through the named fellowships, which took the edge off for the group leaders in terms of taking on someone inexperienced in their field.
Has anyone ever seen bwa samse act weirdly and randomly inserting duplicate lines (whether reads or headers) in the sam file it makes? I'm working with bwa v. 0.7.17-r1188 either base install on my server or on a conda install, and it is being VERY weird with sam file production. samse/sampe makes these corrupt sam files with repeats (either duplicate headers, or will insert read information and then more headers underneath, then more read data), and bwa mem makes "correct" sam files but all reads are unaligned (no matter the sample, for fastas with 2-15 million reads and reference genomes of 3GB). I've tried googling the errors and so far, the best advice I've seen is with the bwa aln+samse route, try manually removing the duplicate lines if there are few of them (which I don't think is my case), or attempting to use bam utils (this fails with the bam conversion too). I'm very confused on how to proceed. Has anyone ever seen this? Maybe with this version of bwa? Is this a bwa dependency malfunctionning? My preference would be to "fix" the software so no weird things happen rather than dealing with the weird sam files on a case by case basis, but I'm really lost right now...
*Thread Reply:* Maria, could you post here a few duplicate lines? Also could you extract the corresponding reads from the fastq-file and post for comparison?
Hey dear community, What tools are you using to make your multiple genomes circular comparison maps ? Anvi’o looks really nice (https://merenlab.org/2016/11/08/pangenomics-v2/), plus I already have some of their stickers, but I was wondering what’s your tools of choice


*Thread Reply:* Plot is from https://sfamjournals.onlinelibrary.wiley.com/doi/10.1111/1462-2920.13015
*Thread Reply:* Vanilla circos ? From scratch ?
*Thread Reply:* yup, but mostly just for final results
*Thread Reply:* Adding further softwares for reference: • BRIG: https://brig.sourceforge.net/ • pyCircos: https://github.com/ponnhide/pyCircos
Does anyone have any information for designing capture probes? I have only captured the human genome and bought a commercial kit. But I guess this is not going to be an option for other species... I am thinking about pathogens (and other animal sp). Thanks!
*Thread Reply:* Depends what pathogen and what animal sp. They all have pretty different genomes etc Any idea what you want to do yet?
*Thread Reply:* @Megan Michel @aidanva @irinavelsko?
*Thread Reply:* It’s all very theoretical right now! Just wondered what the process looked like, if there were specific companies to approach and what sort of information they would need?
*Thread Reply:* I have some code/pipeline (not yet published and definitely not in a "download and run"-state but we can still share it) that takes in a set of (already downloaded) genomes, and will output the unique probes for this set of genomes with a given step size. These can then be post processed to remove highly similar probes (depending on your definitions for this)
It is not the quickest method imaginable but it is robust and usable in my opinion (depending on how many genomes you start with and how big your genomes are, eg if you try to get probes for microbes it is OK, if for eukaryotes it might be too slow).
let me know if you want to peek at the methods section I have written up!
*Thread Reply:* Oh wow Ian!! That sounds great, I would love to have a look if you don't mind! Thank you 😄
Hello all, I was wondering what tools people would recommend to explore recombination in viruses? So far, I have used SplitTree4 on full genomes but the output does not appear to give me details of the possible breakpoints. I also tried to run the same full genome alignments through GARD on datamonkey, but it doesn't load. I was able to make it run going ORF by ORF and it does output possible breakpoints, but I feel I may be making it harder on myself if I go ORF by ORF. Looking in the literature, I found another tool called RDP 5, but it only runs on a PC so as I use Mac, I was wondering if there were any other tools people would recommend (and that are preferably user-friendly)?
Hello everyone, Imagine that I have to do a phylogenetic tree within two weeks for 3 pathogenic species. Do you have any suggestion on a realistic way to do it in a short time? Also keep in mind that I have never done a phylogenetic tree before 😅. Thanks in advance 🙏
*Thread Reply:* Hello Zoé, If it is a phylogenetic tree with bootstrap values that you're looking for, I would personally recommend IQTree: http://www.iqtree.org/ You can set IQtree to look for the best-fit model through the option -m TEST and you can also run the tree using bootstrap approximations (UFBoot and SH-aLRT) which will speed up your run - with the options -bb and -alrt. Of course, if you don't want approximations, then you can use the option -b but your analyses will take longer to run. I always start with the approximations to get a good idea before rerunning the analysis with the other option. The manual is very user-friendly too 🙂
*Thread Reply:* What’s your starting point ? ie. what kind of data/results do you have right now ?
*Thread Reply:* Thank you @Ophélie Lebrasseur I’ll check that 😊 For @Maxime Borry I’m not sure exactly sure what kind of information you need so don’t hesitate to ask for more: So I have “Bacterium 1” that is found in 4 individuals: • KrakenUniq: 160 000 reads, 2 700 000 kmers, after MALT alignment: 93% breadth of coverage • KrakenUniq: 11 000 reads, 330 000 kmers, after MALT alignment: 50% breadth of coverage • KrakenUniq: 8 500 reads, 140 000 kmers, after MALT alignment: 9036 reads and 19% breadth of coverage • KrakenUniq: 5 200, 32 000 kmers after MALT alignment: 6088 reads and 15% breadth of coverage “Virus 1" that is found in 3 individuals: • KrakenUniq: 7 100 reads, 31 000 kmers, visualisation with IGV shows that the whole genome is covered • KrakenUniq: 630 reads, 4 070 kmers • KrakenUniq: 580 reads, 2 500 kmers A second bacterium is found in 2 individuals: • KrakenUniq: 190 000 reads, 81 000 kmers, after MALT alignment: 72% breadth of coverage • KrakenUniq: 2 800 reads, 8 500 kmers, after MALT alignment: 41% breadth of coverage Note: I am rerunning MALT now because I was using the version that had a bug before. But if I understood well, the bug was mostly underestimating the amount of reads assigned to a species so rerunning it should actually improve the result
*Thread Reply:* Do you already have reconstructed genomes (1), or just reads mapped to a reference genome (2) ? • For (1) https://github.com/maxibor/corephylo • For (2) nf-core/eager to generate VCF + https://github.com/maxibor/vcfphylo
*Thread Reply:* Thanks! Is one better than the other? I have the second option, reads mapped to a reference genome
*Thread Reply:* I have done a quick and dirty tree using Snippy to call SNPs and MEGA to build the tree and it works pretty well!
*Thread Reply:* They're just different. You would do (1) if you have de novo assembled genomes, (2) otherwise
Extreeemely dumb question for @Nikolay Oskolkov @Zoé Pochon or anyone on the team… is there a command line download link for the KrakenUniq databases on figshare? Trying wget <https://figshare.scilifelab.se/ndownloader/articles/20518251/versions/1> with the link from the “download all” button but it doesn’t get very far before throwing me a “Stale file handle” error, wondering if that’s at my end or if there’s a better way to do this!
*Thread Reply:* Hi @Shreya, you are doing it right, and the “Stale file handle” error is probably an internet connection break, so it is important to be able to resume downloading. A way to download it is to right click on the download button in figshare, copy the link, then use aria2c to download as you would do with wget. It works the same way, and you could use wget as well, but aria2c can resume downloads if they get interrupted. Nevertheless SciLifeLab Figshare recommends to use their API here https://docs.figshare.com/#file_download, but I have not tried it so can't advise you, perhaps you could test this way as well?
*Thread Reply:* I was also struggling with downloads. Finally, I used aria2 to download separate files. e.g.
aria2c -x 4 "<https://figshare.scilifelab.se/ndownloader/files/37576102>"

Hey everyone, I also have one question which should hopefully be easy to answer. I'm running krakenuniq using the full nt db and I've noticed that it's pretty slow (5h/file even though the files are <1Gb), so I have added the "--preload" flag but I'm not sure it works properly within the for loop. Any ideas why it may run so slow?
for i in $INPUT/merged**; do krakenuniq --db $DB --threads 32 --preload $i --output $i.kraken --report-file $i.report; done
*Thread Reply:* @Lennart Schreiber how much available RAM do you have? Second, the speed of KrakenUniq is not really affected by the input fastq-file but the database size. Third, please check the "--preload-size" option which should be much faster. Finally, the sequential for-loop solution did not really work for me either, so I always try to submit different samples to different cluster nodes
*Thread Reply:* Thank you very much @Nikolay Oskolkov! I allocated a total of 160Gb RAM when I last started it but maybe I should ask for more? I'll try the "--preload-size" option and thanks for the tip about submitting samples to different cluster nodes, I haven't considered that.
*Thread Reply:* If you have 160 GB of RAM and want to fit a full NT krakenuniq DB, I would try "--preload-size 128GB". If it fails (sometimes it does because --preload-size is not very stable yet to my experience), then please try "--preload-size 96GB"
*Thread Reply:* Yes I would also suggest not running it in a for loop but each sample separately
*Thread Reply:* krakenuniq works pretty well for me with the
--preload option
*Thread Reply:* what I do is to first preload the DB and then run the for loop for each dataset and it runs very fast per sample. The limiting step is the loading of the DB. I haven’t tried yet to also use the --preload-size option for the full NCBI DB
*Thread Reply:* Here is the code for an sbatch script:
*Thread Reply:* ```#!/usr/bin/env bash #
SBATCH -o krakenuniqnt.out -e krakenuniqnt.err
SBATCH --qos=normal
SBATCH --cpus-per-task=30
SBATCH --mem=400G
SBATCH --qos=normal
SBATCH --job-name KrakenUniq
krakenuniq \
--db DB-DIR-PATH/ \
--preload --threads 30; \
for file in cat samples.ids; do \
krakenuniq \
--db DB-DIR-PATH/ \
--threads 30 \
--report-file ${file}.tax.report.tsv.gz \
--output ${file}.read.report.tsv.gz \
--gzip-compressed \
--paired \
--fastq-input ${file}.trim.R1.fastq.gz ${file}.trim.R2.fastq.gz ; \
done```
Another not-a-stupid question - for pair-end data, is the universal approach to only use collapsed reads? we're getting some curious / suspicious alignments when looking non-collapsed reads (this was out of curiosity more than anything else) thanks!
*Thread Reply:* Depends on the context. Can be useful for boosting coverage when you've got low endogenous, but you can assume collapsed only reads are more likely to be short and therefore more likely to be really ancient. So in the second context it it maybe a better approach for pathogen screening
*Thread Reply:* So what you're seeing makes sense if you are doing pathogen screening I suspect
*Thread Reply:* exactly, my instinct would be conservative RE the choice of shorter/collapsed reads for ancient pathogens
can see there being contexts of broader inclusion of reads. in this instance the read pairs were often not properly paired (⚠️) but were individually blasting to the genome of interest (spooky). rather stick with the collapsed reads that are more trustworthy ex ante
thanks for sounding that out!

hi all, I have assembled MAGs from ancient metagenomes and would now like to determine wether they are indeed ancient or modern contamination. Here is the code I used to run pydamage (https://github.com/franciscozorrilla/metaGEM/issues/119#issuecomment-1374595930). In short, I concatenate all MAGs assembled from the same sample, then map qfiltered reads to generate a sorted bam file to provide to pydamage.
1) I was wondering if anyone could elaborate on the usage of the -g, --group parameter for the pydamage tool? I have looked through the documentation and paper, but having a hard time understanding what it does or if it is appropriate for me to use here.
2) If I understand correctly, can I then use the output files to determine ancient provenance of each contig by filtering for predicted_accuracy >= 0.67 & qvalue < 0.05?
any comments, suggestions, and/or advice welcomed!
*Thread Reply:* @Maxime Borry would be the best to answer these
*Thread Reply:* Hi @Francisco Zorrilla , 1) the group flag is when you want to analyze at the genome/MAG level, ie: treating all the contigs as if they were a single long contig, otherwise PyDamage analyzed contigs individually. For your use case, I don’t think you’d want to use it. 2) The 0.67 threshold is the one we used for the dataset of the paper. To determine the threshold for you data, you can let PyDamage set it for you with the kneed method (see doc here: https://pydamage.readthedocs.io/en/0.7/CLI.html#pydamage-filter) or decide that a it’s anyway a binary classification program and use a threshold of 0.5 (default) 😉
*Thread Reply:* thanks for the quick response and clarification!
*Thread Reply:* hey @Maxime Borry I was wondering if its possible to use the output of pydamage to try and roughly estimate the age of a MAG/contig?
*Thread Reply:* @Francisco Zorrilla aDNA damage is not only explained by the age of the sequences, but also by a lot of other factors. So it’s unfortunately not possible. AFAIK, the only way to estimate the age of DNA from the sequences alone is the use of time trees (Through ML http://www.iqtree.org/doc/Dating or Bayesian inference https://github.com/Taming-the-BEAST/Basic-tip-dating)
*Thread Reply:* I +1 Maxime's statement. aDNA damage accumulation can be accelerated in warm and wet environments, and slowed down in frozen and dry environments, so it's not a uniform process.
Like Maxime says, the closest you can get is via mutation-rate clock estimates but that requires rather sophisticated analyses in most cases
*Thread Reply:* thanks for pointing me in the right direction! will read more on those methods 🙂
*Thread Reply:* Orlando, L., Allaby, R., Skoglund, P., Sarkissian, C. D., Stockhammer, P. W., Ávila-Arcos, M. C., Fu, Q., Krause, J., Willerslev, E., Stone, A. C., & Warinner, C. (2021). Ancient DNA analysis. Nature Reviews Methods Primers, 1(1), 1–26. https://doi.org/10.1038/s43586-020-00011-0
*Thread Reply:* That might be a good general summary of aDNA
*Thread Reply:* (it's quite broad though)
Hi there! I have a "has anyone else..." or "are we crazy...??" type of question for you all today.
This is something that we keep running into during our decontamination/filtering pipeline of kraken2 output. In many of our samples (calculus of various mammals, humans), among the most abundant classified taxa in our raw kraken2 output is identified as "human". This is particularly weird because this happens even after we remove all reads mapping to the human genome (and host genome for non-human mammals) during preprocessing. Looking more closely at kraken output, we find that the overwhelming proportion of these "human" reads have a split classification, consisting of: 1) kmers that do indeed hit to the human genome, and 2) kmers that do not have a match in the kraken database (persists with kraken runs on larger databases, i.e. full-nt). Typical kraken output for these reads looks like this:
```C MA00605:444:HMGCKDSX3:3:1102:4119:30671 9606 109 9606:12 0:63 C MA00605:444:HMGCKDSX3:3:1103:2401:4554 9606 73 0:3 9606:6 0 C MA00605:444:HMGCKDSX3:3:1104:12138:27712 9606 84 0:38 9606:7 0:5 C MA00605:444:HMGCKDSX3:3:1104:12943:28886 9606 84 0:38 9606:7 0:5 C MA00605:444:HMGCKDSX3:3:1105:9064:8046 9606 89 0:5 9606:3 0 C MA00605:444:HMGCKDSX3:3:1105:9082:8077 9606 89 0:5 9606:3 0 C MA00605:444:HMGCKDSX3:3:1106:19262:1470 9606 65 0:10 9606:3 0:18 C MA00605:444:HMGCKDSX3:3:1107:28583:10848 9606 83 0:44 9606:5 C MA00605:444:HMGCKDSX3:3:1107:20491:32941 9606 119 0:31 9606:1 0:5 9606:1 0:47 C MA00605:444:HMGCKDSX3:3:1107:16260:28620 9606 119 0:31 9606:1 0:5 9606:1 0:47 `````` So an overall classification classification of "unknown" might be more accurate than "human". I've also done a rudimentary BLAST of some reads with these split classifications and some do hit to records of junk/artificial/sequencing artefacts in NCBI.
I've compared this with reads used to classify other highly-abundant oral taxa. While some reads have kmers split with something not in the kraken database, it's nowhere near as prevalent as it is for the "human" reads.
So.... are we indeed crazy or has anyone else come across this before? Do these reads represent artefacts of the human genome we are using (GRCh37), or possibly artefacts involved with the preparation of samples for sequencing? Any suggestions are most welcome, because we usually just hard-code the removal of any "human" records in kraken data before before downstream analyses.
*Thread Reply:* Hi @Adrian Forsythe, what database are you using with kraken2, is that NCBI NT?
*Thread Reply:* Hey Nikolay, I've run these samples through kraken2 using both the "standard" and nt NCBI databases
*Thread Reply:* The presence of human-classified reads in kraken after removing the human-mapped reads is also something we normally observe, but I never invested time to figure out to what they correspond and why they are classified as human. I normally just remove them from the analyses as I consider them confounding…
*Thread Reply:* Ok, good to hear that this has also been observed by others, thanks Nico!
*Thread Reply:* @Adrian Forsythe I confirm that there is sometimes a mess with seemingly "human" reads. I believe, I observed both types of situations, i.e. 1) when kraken does not report as many human reads as I expect, and 2) when kraken reports a large fraction of "human" reads even after a "human reads removal" procedure. I do not have a clear explanation of what is going on but have a few thoughts so far.
- I do not really trust a "human reads removal" procedure which is typically based on mapping a raw fastq to a human reference and filtering away reads mapped with a good quality. Despite this is widely used, this is not a "competative mapping", i.e. the raw metagenomic reads are exposed to only one reference, I have a difficulty with this approach. Exposing metagenomic reads to one reference at a time can be prone to various biases. Other people might think differently @James Fellows Yates?
- NCBI NT has a very poor quality human reference, only small pieces, therefore I would expect a severe underestimating of actual human contamination when using the default NCBI NT unless a good quality human reference has been added manually. In contrast, the "standard" kraken DB should have a good quality human reference.
- Some microbial reference genomes are known to have human contamination https://pubmed.ncbi.nlm.nih.gov/31064768/, so it is a mess

*Thread Reply:* Thanks for the feedback @Nikolay Oskolkov ! If I can add a bit more context:
- I agree, and I should have clarified that, in our preprocessing, the removal of host/human reads is a competitive mapping approach, mapping against a combined reference of human and host genomes (in the case of our non-human samples).
- Yes, this is good point. I would assume then that the amount of "human" assignments would differ between standard and NT databases
- In very few cases, we do find some bacterial species that are represented in the kmer assignments that are split with "human" kmers. I believe that some of these bacteria are even specifically mentioned in the paper you attached
Hi, all. I know that it is common to use Herculase II Fusion to index libraries and PfuTurbo Cx to amplify libraries. Is there a reason people don’t use PfuTurbo Cx for both? Is it because Herculase is cheaper? I have never had dimer issues with PfuTurbo Cx, so I don’t think it’s that.
*Thread Reply:* I believe it's the other way around: pfu turbo to index and Herculase to amplify (ref: https://www.protocols.io/view/illumina-double-stranded-dna-dual-indexing-for-anc-4r3l287x3l1y/v2)
IIRC, you must at least use pfu turbo (or similar) to ensure you 'incorproate' damage into your reads, when you do initial amplification, after which it doesn't matter which polymerase you use. So you can use a cheaper one like Herculase (or a higher fidelity one)
I normally ping @Christina Warinner as my polymerase expert ;)

*Thread Reply:* There’s a paragraph on polymerases under “DNA library amplification” here: https://www.nature.com/articles/s43586-020-00011-0
*Thread Reply:* mmm so perhaps PfuTurbo Cx is used for indexing because it reads through uracil, but Herculase cannot. Then Herculase is used for subsequent amplifications because the uracils are all back to thymines after indexing and Herculase has the same error rate as PfuTurbo Cx, has higher yields, and is cheaper.
*Thread Reply:* Exactly!
Hi all. I have a question related to hybridization capture: what would be the disadvantages of pooling indexed libraries before capture? I am working with indexed libraries that only contain very small amounts of target DNA (hence the capture) so I'm not that worried about "saturating" the baits. As far as I'm concerned, the double-indexing of the libraries should also prevent any contamination/misassignments.
*Thread Reply:* I find pooling as little libraries as possible works best for me (also double-indexed) and the protocol we used suggested adjusting the volume according to which library had more endogenous DNA. This paper talks about the issues with index hopping and also suggest avoiding pooling when the endogenous varies too much. Other may be more useful as I've only been using the arbor RNA-seq baits

*Thread Reply:* Thank you very much for sharing your experience Pooja and also for pointing me to this relevant paper!
*Thread Reply:* We have performed capture on pools of double-barcoded extracts (before indexing), which worked rather well (this was done with museum specimens) https://onlinelibrary.wiley.com/doi/full/10.1111/1755-0998.12699 Our colleagues working with faecal samples make a strong point about determining the endogenous content first (if capturing the host) and pooling samples by endogenous content: https://onlinelibrary.wiley.com/doi/full/10.1111/1755-0998.13300
Hi all! Yesterday at SPAMMTISCH @irinavelsko and I were talking about doing R analysis with Phyloseq and this package offers the opportunity to handle otu tables, taxonomy tables and phylogenetic trees at the same time. However, with Shotgun data we're flying without trees, so we were wondering if anyone has any idea how we could get a phylogenetic tree to integrate into our analysis (for e.g. for phylogenetics-informed diversity studies) with the taxonomy data we get from taxonomic id/classification of shotgun data?
*Thread Reply:* If you use MEGAN you can export nwk trees of output from MALT/MEGAN classfiication
*Thread Reply:* (i.e. RMA6 files)
*Thread Reply:* Thanks for your super fast reply! A couple of people tried this with no success. Also, do you know if there is another way for non-MALT/MEGAN users?
*Thread Reply:* No success in what way?
Not off the top of my head concretely, but I'm pretty sure you can upload litsts of TaxIDs to services like iTol, and/or possibly build something with NCBI entrez... (but don't hold me to that 😬 )
*Thread Reply:* I had no success with the MEGAN-exported trees, and wasn't able to figure out why. It looked properly formatted, but no program would read them. The issue I had with trying to build trees in anything related to NCBI was that they were formatted like the big NCBI taxnomy tree, but not nwk, and I couldn't get the ncbi-formatted trees read by any program to convert them to nwk format. If someone knows how do do that, it might help
*Thread Reply:* Hm interesting, because that's what I did in our pnss paper (the Megan one)
*Thread Reply:* I dunno what the next soaam Tisch will be about but I could try and look what I've done before and report back for them
*Thread Reply:* Yes we definitely need another spaamtisch for phylogenetic trees bc we discussed them only in the last 15 min. However next month I thought we try another format (e.g. inviting an author to talk about their research)😉

Hi all, Has anybody used Recentrifuge or Deconseq to remove contaminating samples from your ancient oral metagenomes? If you have, what samples have you used as negative controls in the case of Recentrifuge or which remove/retain database have you used for DeconSeq? Thanks!
*Thread Reply:* I did not use Recentrifuge myself but my colleagues used sequenced blanks as negative controls when running Recentrifuge (as far as I know)
*Thread Reply:* I have used it, I would say generally that would be blanks or controls but which type usually depends on the study
*Thread Reply:* I've not used the two tools but, as @irinavelsko said at the last #spaamtisch, we used the decontam R package with blanks, but also with archaeological bone samples (femurs) with the package's simple algorithm to additionally remove some environmental contamination too (don't know if that would work for these two tools you mention).
To be honest this would be another good study for someone to do!
*Thread Reply:* Try out different decontamination methods and if they work not just on blanks etc
*Thread Reply:* Thank you all for the replies :)
(just want to say it's really nice to see lots of questions and answers(!) on this channel! Keep it up everyone!)
In the spirit of keeping it up, I have a question!
We are trying to deal with removing PCR duplicates in metagenomic mtDNA data. The situation is where you have reads that have been classified with Kraken as e.g. Bovidae mtDNA reads. These can map to many different mtDNA genomes (cow, goat, sheep, etc), and so you can't necessarily dedup by position. We could just dedup based on sequence identity, but we usually do this based on position. Does anyone have any thoughts? It seems like there must be a best practice..
*Thread Reply:* AFAIK generally in metagenomics people don't really do this, as it's assumed duplication is (mostly) equal across all reads, and it's not really worth the additional computational processing/extra steps to remove them.
I believe people who do do it do it like you said based on sequence identity prior classification (IIRC BBTools has a function for this)
*Thread Reply:* What do you mean by metagenomic mtDNA data though? SG sequenced data against a mtDNA Kraken database?
*Thread Reply:* fastp also does deduplication: https://github.com/OpenGene/fastp#duplication-rate-and-deduplication
*Thread Reply:* Not quite sure what you mean but I have used this to dedup in the past: https://github.com/BioInfoTools/BBMap/blob/master/sh/clumpify.sh But also KrakenUniq is somewhat duplication aware
*Thread Reply:* Clumpify that's the one!
*Thread Reply:* @James Fellows Yates This is capture data, with probes that tile ~250 mammalian mitochondrial genomes. We use Kraken to bin the reads into Family groups (hence Bovidae), and then map to each reference mtDNA genome within that Family (e.g. cow, sheep, etc).
*Thread Reply:* Clumpify looks good - assuming that it still requires reads to be the same length, then allowing distance between reads would help catch sequencing errors.
*Thread Reply:* We also thought about just not deduping. How then would you calculate the number of fragments that you have as data? e.g. 100 DNA fragments mapping to a genome is a lot more data than 10 fragments with a duplication rate of 10.
*Thread Reply:* Ah I see. Well that isn't metagenomics really although you used the tool upstream, you're asking a slightly different question.
You shouldn't really have sequencing error at this stage, you should have QC'd your reads to remove badly called bases by the sequencer.
And really in metagenomics we don't care necessarily about the exact number of reads but rather relative abundance so it's also why we don't tend to need to dedup (with the assumptions etc).
*Thread Reply:* Well by metagenomics I mean stuff like Microbial ecology/microbiome etc
*Thread Reply:* Are you gatekeeping metagenomics, James?? 🙂
*Thread Reply:* It's a bit different with e.g. pathogen detection, but again the metagenomic part is too detect, afterwards it's standard Genomics usually
*Thread Reply:* Blame reviewers for being picky with terminology!
*Thread Reply:* But no, it's more have to think a bit differently
*Thread Reply:* I guess I think of it as metagenomics because our libraries do have multiple species in them.
*Thread Reply:* Ah but then all biological samples are metagenomic ;)
*Thread Reply:* (because they are sediment samples)
*Thread Reply:* But anyway, back to the question: I think clumpify/fastp will help
*Thread Reply:* But in terms of reporting it depends what your reviewers would expect as their preferred numbers
*Thread Reply:* I would assume at this stage it would be standard Genomics so the 10 reads bit ?
*Thread Reply:* My only other suggest that you could try something along the lines of: map two one mtDNA genome, dedup on that, then re'map' back to the 'multi reference'
*Thread Reply:* Although it's not going to be perfect
*Thread Reply:* Btw: "It is designed for accurate data, meaning Illumina, Ion Torrent, or error-corrected PacBio; it will not work well with high error rate data. Even for Illumina data, quality-trimming or error-correction may be prudent."
From clumpify
*Thread Reply:* And "For both SE and PE data, fastp supports evaluating its duplication rate and removing duplicated reads/pairs. fastp considers one read as duplicated only if its all base pairs are identical as another one. This meas if there is a sequencing error or an N base, the read will not be treated as duplicated" from fastp
:/
*Thread Reply:* So I guess if you're concerned about that, this will remain an issue from then normal ways I'm aware of
*Thread Reply:* > fastp considers one read as duplicated only if its all base pairs are identical as another one Yeah, that's what I'm concerned about, but it seemed like clumpify dealt with that? Even if not, there has to be some sort of hacky way to do what I want, I agree.
*Thread Reply:* e.g. in the clumpify options:
```subs=2 (s) Maximum substitutions allowed between duplicates.
subrate=0.0 (dsr) If set, the number of substitutions allowed will be max(subs, subrate**min(length1, length2)) for 2 sequences.```
*Thread Reply:* Although there does seem to be some hacky-ness even to that solution, given this additional option:
scanlimit=5 (scan) Continue for this many reads after encountering a
nonduplicate. Improves detection of inexact duplicates.
*Thread Reply:* Yeah exactly...
*Thread Reply:* @Benjamin Vernot I agree with @James Fellows Yates that it is not crystal clear whether one should do deduplication in ancient metagenomics or not (I am talking now only about "microbial metagenomics", it might be different if one is after mammalian reads from an environmental or sedaDNA).
My understanding is that the concept of duplicate removal comes from the whole-genome sequencing of a sample with a clear host (human, animal etc.). There, we know that the host DNA is definitely present in the sample, we often map the reads to the host reference genome alone, and all we want is to do a good variant calling for a popgen analysis, and duplicates can obviously bias the variant calling, therefore they are removed.
In contrast, when working with ancient microbiome, we generally have very little prior knowledge about what microbes are present in a sample. I.e. accurate variant calling is not our first challenge, we are often satisfied and happy if we reliably can conclude presence of a certain microbe in a sample. Therefore we are working simultaneously with thousands (or even millions) reference genomes for an unbiased profiling, and our major first challenge (in my opinion) is the microbial discovery itself (yes or no, present or absent), and not really the genetics of the microbes. And not even quantification of microbial abundance (at the beginning). For doing microbial profiling of ancient metagenomic samples we are not really after good quality alignments immediately. Therefore, I doubt deduplication would be beneficial for microbial discovery (in contrast, it might reduce sensitivity of the discovery), and I also doubt duplicates affect dramatically even microbial abundance quantification. Think about RNAseq, where there is uneven coverage across genes (very similar to metagenomics uneven coverage across organisms), people do not generally do duplicate removal in this field, because variant calling is not their major goal, and duplicates do not really bias the discovery (expressed or not) and gene expression quantification.
However, once a microbe has been reliably detected, one can of course proceed with all the steps from popgen analysis involving good quality mapping to a good quality reference (now there is only one reference!), deduplication, filtering poor sequencing quality or poor mapping quality reads, masking certain regions etc.. This all is needed for a good variant calling.
In summary, there is a difference in motivation behind duplicate removal when working with single and multiple genomes.
P.S. I certainly confirm that removing duplicates based on sequence identity along is not optimal. A definition of a PCR duplicate (to the best of my knowledge 🙂 ) is two reads with identical sequences and identical start and stop positions of mapping. The problem here is that the start and stop positions are very computationally expensive to compare across all reads, therefore some common duplicate removal programs (such as samtools rmdup and Picard) use some heuristics and approximations (for example, they compare only the start but not the stop positions) which result in tones of non-duplicates being removed and tones of duplicates remained. I have seen a lot of artifacts of duplicate removal from those two programs but can't say anything about fastp and clumpify.
*Thread Reply:* Thank you @Nikolay Oskolkov - that explains much better than my bad-late-evening rambling 😅 (looking back now I do see how it could see I was maybe 'gatekeeping' but that was not at all the intention - sorry @Benjamin Vernot 🤦 - Nikolay puts it in a much better way)
*Thread Reply:* Maybe another way to go about hte question: what is the precise need to dedup exactly? One could argue given you have similar genomes, does it really matter if they are randomly distributed (they aren't informative for variant calling etc)?
*Thread Reply:* Thank you both for your thoughts!
*Thread Reply:* I guess I'm not fully on the train of "no deduping necessary," even for microbial quantification / detection. For example, your initial ancient DNA fragments will not all be amplified evenly, and so if you don't dedup you'll favor reads with a "better" GC bias, which could in turn bias which microbes you detect. And my understanding of RNAseq is that you don't dedup because you expect many truly independent reads to have the same start and stop, simply because of how the data is generated / exon structure.
*Thread Reply:* But I don't have a strong analysis to back that up, it's really just my gut, and I trust you when you say that people don't typically do it!
*Thread Reply:* I think everyone would agree with you that it's not not necessary, but it's not a priority problem 😉
*Thread Reply:* Yes, that makes sense!
*Thread Reply:* For us, the reason we would like to dedup is that we use the number of molecules observed for a particular species as a sort of significance test. i.e., if we observe 10 molecules, maybe we don't really trust that species was present, but if we see 100, we do.
*Thread Reply:* So we want to distinguish between 100 truly independent molecules, and 10 independent molecules with a duplication rate of 10.
*Thread Reply:* .. and we want to do this in a systematic way
*Thread Reply:* Ok! And how does the sequencing QC look (e.g. in fastqc)? Do you have a big concern about base-calling quality resulting in miscalled bases? I ask as after trimming nowadays from my data these look very clean.
Because my feeling then just pulling out the reads IDs Kraken report as bovidae, running a start/stop location with exact sequence match would be sufficient in this case, particularly as you're dealing with very short reads with both ends sequenced (I presume)?
*Thread Reply:* I agree that generally deduplication should be a more conservative way for both detection and variant calling.
I guess a part of the problem is also that we often work with shallow sequencing data in ancient metagenomics and do not want to throw away a large fraction of our reads. When working with a single reference genome, people can afford doing deduplication for clear reasons (i.e. good variant calling). When working with multiple reference genomes, one should probably think twice whether deduplication is not unnecessarily conservative. The risk is that with a thorough deduplication we might discover very few microbes. They will be very, very reliable but most likely modern contamination 😂
*Thread Reply:* TBH I'm not really sure what the FQ looks like for these libraries, but we regularly get duplication rates in the 20s, and yet have very sparse coverage of the mtDNA genome. Even with a low error rate, you'd expect some amount of those duplicates to have mismatches.
I agree that the way to go is to just dedup mapped sequences, we just have this (apparently unique) issue of mapping to multiple genomes. But I will figure out a way around it! 🙂 And that way around it might just be to ignore the issue and have a handful of duplicates kept around. Thank you guys for your help!
@Nikolay Oskolkov I have to admit that I don't follow your argument. If you have very shallow sequencing, then deduping is more robust, and less likely to remove truly independent reads. Keeping more duplicate reads around doesn't give you more signal!
*Thread Reply:* > Keeping more duplicate reads around doesn't give you more signal! For some tools it does, as they do hard cut offs for minimum reads (e.g., drop evertything under 10 reads, rather than a percentage of e.g. 1%)
*Thread Reply:* But ultimately yes - I think it's a tricky problem! I have had that issue with multi mapping but not at such low such shallow coverage I guess 😕
*Thread Reply:* But then..... why not just duplicate all your reads so they don't get removed, or change the threshold? The thresholds are there for a reason, right?
TBH, this sounds like using a particular statistical test because it gives you p<0.05, and not because it's the right test! I don't mean that is what you're talking about doing, but I"m just saying why my mind is resisting it 🙂
*Thread Reply:* > But then..... why not just duplicate all your reads so they don't get removed, or change the threshold? The thresholds are there for a reason, right? Some people do in fact do that (Alexander Herbig's group in some contexts!), but the tools I'm referring to are often designed for modern metagneomic samples, not very borderline samples that we deal with (e.g. for pathogen detection), where later you would go capture etc. etc.
*Thread Reply:* Yeah, if you're going to capture for further validation, then it makes sense to me to take even very borderline cases. I guess in those cases I would still rather have deduped reads, because then I know how much data I have.
*Thread Reply:* But... I think we could go back and forth on this forever 🙂
*Thread Reply:* I"m going to go play UNO now 🙂
*Thread Reply:* Tbh much better use of time 🤣
*Thread Reply:* I thought so too, but now my son is pitching a fit because I won! ¯_(ツ)_/¯
*Thread Reply:* Hmmm that sounds very very familiar with my evening two days ago... Have you tried dobble? That can also be... Emotional
*Thread Reply:* We haven't, I'll have to check it out. He's just getting into games a little bit, so we've really only done Memory and UNO.
*Thread Reply:* Oh, I have seen dobble! It seems pretty cool.
*Thread Reply:* Ah ok! Doubble is between those two in complexity so he might enjoy it!
*Thread Reply:* Is this one where there is a component of physical prowess / speed?
*Thread Reply:* Like you have to recognize you have a match, and be the first?
*Thread Reply:* Yes exactly!
*Thread Reply:* @Benjamin Vernot @James Fellows Yates I agree that a detection threshold can be adjusted to be sensitive enough after duplicate removal has been applied. If I was sure that deduplication programs do a good job, I would probably do that. However, as I mentioned previously, I have seen too many artifacts with Picard and samtools rmdup / samtools markdup, and I know that people, who have seen this as well, write their own deduplication scripts to be on a safe side. This still, however, does not mean that we should not deducplicate provided that we have a good way (accurate and computationally efficient) to do it 🙂
There is another problem here that I call a "single read limit". Imagine, after deduplication, you found 2 reads in your sedaDNA sample that really look like mammoth reads. I mean, the two reads stand the competitive mapping against a bunch of other mammals (i.e. they have a higher affinity to mammoth compared to other mammals), and they have mammoth-specific alleles that are not present in other mammals. Would you follow up this hit with a capture? Maybe. However, what if, in addition to the 2 mammoth reads, you see 100 Bovidae reads, 5 wolf / dog reads, 20 cat reads, and 1 bison read? I believe you recognize the situation 🙂 What hit would you follow up? Are they all ancient? Well, hard to say for sure for 2 mammoth reads, we can't do any meaningful stats on them 🤔 But are they mammoth reads at all? Are we sure that, what we thought was a mammoth-specific allele, is really never encountered in a cow? How confident are we that an accidental damage, sequencing error, misalignment, poor quality reference, glitch of nature and other technical factors, have not affected our conclusion about absence / presence of a mammoth DNA (based on 2 reads only) in our sample? Do we really have multiple mammalian organisms in our sample, or all those reads in reality originate from a single mammal? Now, imagine that you have hundreds samples and hundreds mammals, plants, birds, fish, microbes etc. with only a few reads assigned / mapped to their references after deduplication. You should have some statistical approach for processing / digesting those numerous hits (unless you have time to visually go through all of them in IGV). I am not saying it will be a super-correct statistics to work on a large number of non-independent reads (if you skip deduplication), but at least your statistical algorithms will not fail technically. If you, however, have just a few reads after deducplication, there is no way you can get any meaningful stats from them (unless you are a Bayesian 🙂 ).
This all does not mean that we should not deduplicate 🙂 I just want to say that deduplication is not our major problem 🙂 And therefore, I believe, is not widely emphasized in the metagenomics community. Because it is not a crucial factor that affects our decision a lot; as you said, the threshold can be adjusted. You can deduplicate if you want or you can skip it, I doubt it is going to be decisive (for detection). However, deduplication might bring technical statistical problems with handling too few reads. In contrast, when working with a single genome, PCR duplicates can really bias all the e.g. selection stats, so a good deduplication is one of the major problems there.
I would say, a truly unbiased competitive mapping against numerous good quality references is the major computational challenge in ancient metagenomics that is not really solved (in my opinion). Once it becomes feasible to fit good quality references of hundreds of mammals (and other organisms) into the memory, for an unbiased competitive mapping, we could revisit the deduplication issue 🙂
*Thread Reply:* Just came across another tool that may do it: https://telatin.github.io/seqfu2/tools/derep.html
Is anybody aware of a published study of ancient oral DNA in a dilution experiment? Something like what they tested on the decontam paper but with ancient oral DNA?
*Thread Reply:* Nope. I know very few aDNA microbe methodical papers actually
*Thread Reply:* Thanks @James Fellows Yates
Hey all! For people using kraken2 on ancient microbiome samples: what is your go to confidence level? I've read some articles online and they seem to suggest that it might be better to use 0.1 (rather than the 0.0 default). For example see https://github.com/DerrickWood/kraken2/issues/265. But then this is based on modern DNA. I've been trialling some values 0.0 vs 0.05 and 0.1, but I find that if I use 0.1 the classified reads are quite limited, and I thought that it might be a bit too strict for aDNA?
*Thread Reply:* Hi Meriam, I've been asking myself the same, and have been playing around with values up to 0.2
Good morning all, I have a Beast2 question - does anyone know if it is possible to get the posterior distribution of the TreeHeight of an internal node? And if so, would anyone have an idea of how to do it? I've been looking online and on the beast.user google group but I haven't yet found how to do. Any help would be greatly appreciated 🙂
*Thread Reply:* I believe @Arthur Kocher @aidanva used/have used BEAST2, also likely @Meriam Guellil
*Thread Reply:* If I remember correctly, you can achieve this by creating a taxon set that contains all the sequences for which this node of interest is the MRCA (see p. 24 of this tutorial: https://taming-the-beast.org/tutorials/FBD-tutorial/FBD-tutorial.pdf). Once you have created this taxon set, you can set wide priors to the dates of this taxon sets and let BEAST sample them the posterior. The tutorial is a bit extensive and there might be a more elegant way, but I remember doing this in BEAST1 for my master thesis a long time ago.
*Thread Reply:* Thank you so much @Alex Hübner 😊 I'm going to give it a try and report back here!
Is there a consensus (or personal strong opinions) on estimates of alpha diversity? We'd like to do it through time, so counting on things evening out with many samples is not possible, as we have an evenly spaced temporal dataset with only a few samples for each time point. Have been looking at the breakaway method to correct for undetected taxa (https://rss.onlinelibrary.wiley.com/doi/full/10.1111/rssc.12206) but could not find a good/complete implementation. Any and all recommendations are welcome!
*Thread Reply:* not helpful at all, but I was always a little skeptical with alpha diversity when it comes from ancient samples because contamination will skew that like crazy (and I don't think there is a good environmental decontaminatiom tool).
*Thread Reply:* @irinavelsko do you have any thoughts?
*Thread Reply:* Agree on the issue of contamination but right now more focusing on the effects of different sequencing depth and how to control for it without going through rarefaction (again, for a cross-time comparison, where it is impossible to group samples and hope that things even out)
*Thread Reply:* My experience with alpha diversity measures leads me to believe we need to do some testing with simulated datasets to be able to understand how this affects different metrics
*Thread Reply:* Which I know isn't actually helpful, but maybe you have an eager masters student?
*Thread Reply:* I did some testing in both 10.1128/mSystems.00080-18 and the supplement here https://doi.org/10.1093/pnasnexus/pgac148, but something thorough and systematic wold be really helpful
*Thread Reply:* Thanks a lot, @irinavelsko. Do you or any others have a take on the breakaway method? It sounds quite good and seems to be well-used, just not in ancient metagenomics.
*Thread Reply:* I haven't heard of it before, but I'm interested to know how it works for you if you test it
*Thread Reply:* @Adrian Forsythe is testing it right now, so hopefully we'll be able to report back
Hello 🙂 can anybody share the “sample_age.txt” we used for Beauti/BEAST during the summer school?
*Thread Reply:* If it was provided by Aida and Arthur, it should be in here: https://zenodo.org/record/6983185#.Y9BUM6TTUoA
*Thread Reply:* All the material is on the website, under the 'walkthrough' tabs
*Thread Reply:* https://spaam-community.github.io/wss-summer-school/#/2022/day-5?id=walkthrough
*Thread Reply:* yes, we inclueded within the files. If you have any questions about it let. me know Maria 🙂
*Thread Reply:* I found them!!! thanks Aida!
*Thread Reply:* I see how it is. I don't send you enough cute dog videos, is that what you're saying?
*Thread Reply:* It's my raison d'être 😁
Hi all! has anyone had experience using Diamond for taxonomic profiling of ancient datasets with lots of very short reads (<90 bp)?
*Thread Reply:* Hi @Guillermo Rangel, I did 🙂
*Thread Reply:* Hi Nikolay, thanks for replying 🙂 and how did it go for you? I’m currently trying to run different profilers on my datasets using no-core/taxprofiler….. At the moment I’m running Kraken2, Krakenuniq and Diamond, but I’m currently having issues to finish the Diamond runs, or at least in a decent amount of time (I’ve got a dataset with ~480M reads that crashes after 2 days running, and the log file doesn’t really tell me much about it, perhaps a memory issue?). I was thinking of setting the --fast option but I’m afraid it’ll affect the sensitivity?
*Thread Reply:* Yes, DIAMOND is much slower than nucleotide-based classifiers (just think about the additional cost of translating the nucleotide sequences to protein sequences), and will probably not classify the vast majority of your reads. On the other hand, DIAMOND can be more conservative / robust in its classification compared to Kraken, i.e. less sensitive to noise. From my experience, DIAMOND results almost always agreed with Kraken results. However, DIAMOND is a modern metagenomics thing which is more suited for long reads, while for ancient metagenomcs DIAMOND is probably not optimal.
*Thread Reply:* Hi @Guillermo Rangel, I am not familiar with your error message that you get when running DIAMOND, but I second Nikolay about that you should be careful and check that your samples have roughly the same DNA length distribution. Due to the translational step, DIAMOND is not really able to align anything specifically that is shorter than 20 amino acids (or 60 nucleotides). Therefore, an excess of sequences < 60 nucleotides in a sample compared to other samples will give you skewed profile because the short reads won’t be properly aligned.
*Thread Reply:* My rule of thumb is that I consider Kraken as a fast version of blastn while DIAMOND for me is a fast version of blastp 🙂 Regarding the technical limitations, I think I confirm that I had much more headache with DIAMOND than with Kraken. So my suggestion would be to skip DIAMOND at all if you are experiencing technical problems, Kraken results should be good enough, I believe 🙂
*Thread Reply:* thanks you so much @Nikolay Oskolkov and @Alex Hübner for your answers. Indeed, my samples have lots of reads below 60 bp…. I was actually also thinking about the issue of aligning the rather short peptides that would be translated from these very short reads… so I think your suggestions make total sense to me… ok, so I’ll stick to Kraken 🙂
*Thread Reply:* Agreed, I wouldn’t recommend using DIAMOND for ancient DNA
*Thread Reply:* thanks @Christina Warinner 🙂
Hello all, I have another BEAST2-related question. I am trying to run a relaxed clock (log normal) on a dataset (full genome, no partition) with tip-dating. I have followed the following tutorial https://www.beast2.org/2015/06/23/help-beast-acts-weird-or-how-to-set-up-rates.html in order to understand how to best set the 'substitution rate' in the Site Model panel and 'Clock.rate' in the Clock Model panel. For the former, I have kept it as 1.0, no estimate. For the clock.rate, I have inputted a starting rate based on the values I found in the literature. By clicking the pencil next to the estimate box, I've also been able to add a lower and upper boundary and tick the estimate box. What I don't understand is why the 'estimate' box for the clock.rate itself is ticked but remains greyed out? Does anyone know what this greying means? When I run Tracer based on the results, I do have the ucld.Mean estimate but I'm unsure if it's taken my parameters into account. Alternatively, I have seen you could click on Mode > Untick Automatic Set Clock Rate, in which case the box becomes ungrey, but I am unsure that's necessary based on what I've read in tutorials and beast user groups. Essentially, I don't know what I am supposed to do 😅 I can run two analysis and see if they differ in the results, but if anyone has any clue, I would love the insight 🙂 Thank you!
*Thread Reply:* So I found the answer to my own question - I'm posting it here if it's of any use to people - it's essentially because I am using tip-dating, I have strong priors and therefore, Beast automatically detects that it can be estimated. Pretty straightforward but it took me time to get my head around it 😅 Also, this blog post may be useful: https://www.beast2.org/2020/05/20/estimate-box.html

Hey everyone! So, to follow up on Ben Vernot’s previous post we looked into using clumpify, and ran into a problem that maybe someone here has experienced or knows how to remedy.
In short, I am looking at mtDNA of mammals in sediments captured through a mt mammal capture probeset. The sequenced reads were then assigned to specific taxa with kraken – here we’re looking at all reads assigned to a family (i.e. Canidae, Bovidae) or to anything “below” that family, i.e. Canis familiaris. We then map those reads to the mtDNA reference genomes for everything in the family. For example for Canidae, I have reads mapped to dogs, coyotes, wolves, etc. In the file, there are PCR duplicate sequences that I want to collapse into one and have been trying to find the best way to do this.
To test, we’re starting with a bam file of 2399 reads that have been classified as Canidae by kraken.
We have tried a few things so far:
- When we just get unique DNA sequences (with uniq), we get 799 unique sequences.
- When we look at mapped positions (with samtools rmdup), we get 1344 unique sequences. The reason this is larger is that some duplicates map to multiple Canidae reference genomes, and so identical sequences often don’t have the same position.
- When we use rmdup followed by uniq on the sequences, we get 775 unique sequences.
- When we use clumpify with the dedup parameter, we get 788 unique sequences, which seems pretty good, but some of these are clearly PCR duplicates with 1-2 edits between them.
I then tried messing with other options for clumpify (the kmers, substitutions, and border parameters) and s10, k5, b0 seems to be the “best” option, reducing the number of sequences to 772.
This is the command line: clumpify.sh in=examplefasta.fa out=examplefasta_clumped.fa dedupe=t scanlimit=2500000 subs=10 k=5 border=0 ignorebadquality
However, in looking at the file there are still two sequence pairs that have only one mismatch between them. I made a new fasta with just those four sequences to see if maybe it was something with the file, but clumpify is still not collapsing them.
This is the file with the sequences I have that are not working:
>test1 TATGGGGTCAAAACCACATTCGTAGGGGCTTGTCTTGCCTGT >test2 TATGGGGTCAAAACCACATTCGTAGGGGCTTGTCTTGTCTGT >test3 TTTATGGGTAGCTCGTCTGGTTTCGGGGAGCTTAGTTTAAGTTCTTTGTGTTAAGTTGTTT >test4 TTTATGGGTAGCTCGTCTGGTTTCGGGGAGCTTAGTTTAAGTTCTTTTTGTTAAGTTGTTT
When running clumpify, the output is odd as it not only does not collapse them but it also rearranges them in a way that moves two of the like sequences away from each other.
>test3 TTTATGGGTAGCTCGTCTGGTTTCGGGGAGCTTAGTTTAAGTTCTTTGTGTTAAGTTGTTT >test2 TATGGGGTCAAAACCACATTCGTAGGGGCTTGTCTTGTCTGT >test1 TATGGGGTCAAAACCACATTCGTAGGGGCTTGTCTTGCCTGT >test4 TTTATGGGTAGCTCGTCTGGTTTCGGGGAGCTTAGTTTAAGTTCTTTTTGTTAAGTTGTTT
Does anyone know what could be causing this or if I am missing a parameter in clumpify that would resolve this? I have only done this for one library that I have and I want to resolve this before running this on the others.
Even if nobody has seen this before, maybe this exploration will be helpful for someone. Thank you for your help!
*Thread Reply:* Looking again at the bbtools docs, I wonder if dedupe might actually be a better tool: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/dedupe-guide/
*Thread Reply:* Dedupe has 6 phases, most of which are optional and depend on the processing mode. They are always executed (or skipped) in the same order.
1) Exact Matches.
During this required phase, sequences are loaded into memory, and exact duplicates (including reverse-complements) are detected and discarded. Hashtables are filled with sequence hash codes of sequences. If containments or overlaps will be examined in later stages, kmers from the ends of sequences will also be added to hash tables. After this phase, the input files will not be used again.
*Thread Reply:* clumpify seems to be doing something slightly different with the actual merging of clusters being an 'extra'
*Thread Reply:* The reason why I say that is I see this: . The clusters are not guaranteed to be overlapping; rather, they are guaranteed to share a kmer, meaning they are likely to overlap.
*Thread Reply:* So maybe that's why some of your reads are moved away from each other
*Thread Reply:* Ahhh okay, that’s interesting. I am going to mess around with BBtools dedupe instead of clumpify and see if that resolves things
*Thread Reply:* Wuld be curious to knw if it works 🙂
*Thread Reply:* I’ll let you know how it goes ☺️ Thanks!

Hi everyone, I am using FigTree to visualize a tree created with beast2. I am trying to understand the section for node labels, but I cannot find a good explanation for the options. What essentially is the difference between "CAheight95%HPD" and "height_95%_HPD"? Anyone used that recently? What options did you choose? Thanks for the help! 🙂

*Thread Reply:* Hi Magdalena! :headbangingparrot: So the node label annotation depends not on figtree but originates from the tool with which you created the tree. I guess you should check the documentation of that tool. Figtree just displays it as colour/label/whatever. How did you create the trees?
*Thread Reply:* Today at 5pm (German time) there is the informal #spaamtisch meeting, talking also about phylogenetics today. If you have time to join, feel free to discuss the tool there.
*Thread Reply:* Hi Jasmin! Thanks for your reply. I created the tree with beast2 and checked the documentation. I did not find any good explanation on the node labels yet, however, I will endeavor to keep trying 😉
*Thread Reply:* I've used beast (+figtree) recently, and used CAheight95%HPD for plotting (that's what others in our group use), but am also not clear on the difference between that and height - in the vast majority of cases they're identical, but I have had a couple of cases with slight differences? would also be interested in finding out what the difference is 😅
*Thread Reply:* Heya, I found the following on the beast user group: "The CA should stand for "Common Ancestor" and it's 95% height should be different from just height95%HPD (unless the clade has pp = 1) because the clade age estimates are based on ALL posterior trees, and not just the subset for which a particular clade was monophyletic. The Common Ancestor Tree (CAT), which is estimated with treeannotator by invoking the "-heights ca" option is an effective way to deal with negative branches, which is a product of nodes in the MCC tree being older on average than the average age of the immediate parent node. You can find more details in Heled and Bouckaert 2013: https://bmcevolbiol.biomedcentral.com/articles/10.1186/1471-2148-13-221"

*Thread Reply:* Wow, cool thanks a lot! 🙌
Hi All,
I am interested in using MaAslin2 for performing a differential abundance test. This program has several options for normalization and analysis method. I was curious if anyone has any thoughts on which combination I should apply to my ancient microbiome dataset. By default, the program uses TSS for normalization and a linear model for analysis method. I was thinking of using CLR and LM but not sure if this is the most appropriate. If people have thoughts about this or recommend that I use another DA tool, I am all ears.
Thanks for the help!
*Thread Reply:* Hi Sterling, I am not familiar with MaAslin2, but your suggestion about CLR + LM looks good to me. I personally, for any differential abundance test (whether it is microbial abundance or e.g. methylation, gene expression, pixel intensities etc.) would go for a non-parametric Mann-Whittney U test, as the most robust to my experience, rather than any other fancy method. However, if you have confounders to correct your differential test for, this would probably be easier to do within a linear model. Nevertheless, a linear model typically has unrealistic assumptions on residual distributions, this is a drawback of this approach. In summary, to be as conservative as possible I would go for a Mann-Whitney U (MWU) test, but if you have clear confounders, they should be regressed out prior to the MWU test 🙂
*Thread Reply:* Thanks Nikolay! And sorry for not mentioning this in the previous post. One of the main reasons why I have turned towards MaAslin2 is because I can incorporate fixed and random effects (i.e. confounders) into a single model, whereas I cannot do this with others like ANCOM-BC or Lefse. Out of curiosity, do you have any experience with CPLM (Compound Poisson Linear Models)? I have been trying to do a literature search on this but haven't found anything that is useful.
*Thread Reply:* CLR transform + linear model is also my recommendation: you get a lot more flexibility than any other solution
*Thread Reply:* and if you have random effects, just switch to a linear mixed model
*Thread Reply:* a nice review, with code examples in the linked gh repo https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04193-6

*Thread Reply:* Thank you for this!
*Thread Reply:* @Sterling Wright regarding CPLM, I do not know what it is but I know what Poisson linear model is and assume (from the name 🙂 ) that CPLM might be some mixed model (or bayesian hierarchical model) extension of the Poisson linear model. So, no quick help for you, but I am ready to keep discussing this if you have specific questions. Perhaps I can read up on CPLM if you want to dig into details
*Thread Reply:* I would love to discuss it further but only if you’re interested and there would be some benefit to you. You have a much deeper understanding on this than me. However, I can also try to reach out to my statistics department if you overwhelmed with other commitments.

Hello everyone!
This is a question for all of you who work with hard boney samples! Currently, in our lab, we process bone and teeth samples using a hand drill type Dremel 3000-N/10. However, after processing some samples (and all the cleaning) the bone dust has apparently gotten into the inner system rendering it useless. We are debating whether to repair it or buy another one, so I'd like to know what other type/brands of drill you use or if you have any advice on how to deal with this!
Thank you very much for your help!
*Thread Reply:* I have no experience myself, but according to our protocols we use something from KAVO
*Thread Reply:* @Gunnar Neumann or @Alina Hiss might know and have advice? I believe they've done a lot of sampling
*Thread Reply:* Yes. We use the one indicated in the protocol. Good question though about powder getting inside the system. My impression is that these are quite sealed enough to not have this issue. You can take off the tip for easy cleaning inbetween samples and it has this little rubber ring at the tip which should also prevent the powder to get inside the drill. (Very easy to get lost in the drain though so better order some spare ones). We still have to send the drills in for repairing sometimes, but I admit I don’t know the reasons here. All done but our technicians …
*Thread Reply:* I’d recommend getting a foot pedal operated dental drill. Much safer, less contamination risk, and no air blowing near the sample because the motor is separate from the cutting tool.
*Thread Reply:* I’ve worked with different tools in the past. If you stick to Dremel I would definitely recommend using the “Welle” (I don’t know the english translation, here the link: https://www.dremel.com/ch/de/p/dremel-biegsame-welle-26150225ja) ideally with a foot pedal. I’ve also used an older dremel “Welle” which is difficult to clean, in this case I wrapped it with aluminum foil after fixing the drill bit.

*Thread Reply:* For the Bern lab I recently ordered the KaVo Ergo-Grip tool (with a pedal as well), after comparing different models together with a colleague who happens to be a trained dentist. We picked this one because it can be used in both directions (forward and reverse): https://www.kavo.com/dental-lab-equipment/kergogrip-laboratory-handpieces We didn’t get it delivered yet, so I haven’t tested it yet.
*Thread Reply:* I used the Proxxon MICROMOT 60/EF in Copenhagen, with the footswitch/pedal. https://www.proxxon.com/en/micromot/28500.php. It’s very durable
*Thread Reply:* Thank you a lot for everyone's response. We are looking thoroughly through the options. My supervisor and I are deeply grateful! ❤️

Good day. Anyone knows if there are any accessible lists of archaeological sites online? I am looking for Tirup village site in Denmark and i’ve gone as far as translating Danish reports, trying my best to not flip over google’s suggestion to search for it in Sweden and trying to navigate amongst articles about Trump visiting cemeteries and other nonsense. So, even a location for this specific site would be helpful actually, if there are no available.
*Thread Reply:* Unfortunately not, you have you do triage based on literature
*Thread Reply:* So follow up citations in the paper for the archaeological report and see if they have maps etc
*Thread Reply:* Normally research gate can help with this
*Thread Reply:* Let me know if you need a better Danish translator than Google!

*Thread Reply:* Tirup isn't a village, it's just the name given to the site which was located in west Horsens, Jutland. The site no longer exists as it's now an industrial area. The exact location is below (with some more information, in Danish). Let me know if you need any help with translation. Tirup kirketomt
*Thread Reply:* Thanks a lot. I was wondering if Horsens have spread over the location to be fair. Boldsen in 2000 nicely described surrounding areas, but one can’t help it if it’s just not there, between the two places he says it should be. Anyway thanks a bunch for the precise info.
*Thread Reply:* @Bjorn Bartholdy I’m using https://www.deepl.com/translator which is somewhat better even with a free version. So the text made sense. 🤓
*Thread Reply:* Oh cool, haven't tried that one!

Hi y'all, I am trying to combine two the two SILVA databases for SSU and LSU for a malt screening (inspired by https://doi.org/10.1038/s41467-022-33494-4 "Ancient marine sediment DNA reveals diatom transition in Antarctica") I have concatenated my two reference databases into one but I am uncertain how I can create a mapping file or if there is a malt-build option that allows me to use two mapping files. I cannot fild the answer to that in the paper. I tried a bit naive with ./malt-build --input path/SSU_LSU_comb/SILVA_138.1_SSU_LSU_Ref_Nr99_tax_merged.fasta.gz --sequenceType DNA --index path/SSU_LSU_comb/index -s2t path/SSU_LSU/malt/SSURef_Nr99_132_tax_silva_to_NCBI_synonyms.map.gz -s2t path/SSU_LSU/malt/LSURef_132_tax_silva_to_NCBI_synonyms.map.gz -v Does anyone have a suggestion? best, Freya
*Thread Reply:* You would need to combine your mapping file together as well
*Thread Reply:* Conceptually anyway
*Thread Reply:* I've never tried making my own mappung file.
*Thread Reply:* Thanks, I will try that, I just imagined that duplicate sequences might become a problem
*Thread Reply:* I think it should be ok if the taxonomy IDs are the same for the same species in both
*Thread Reply:* well, something is running. Thanks a lot 🙂
Non-ancient q, is there some sort of database containing nucleotide sequence data for the isoforms of a given gene across different strains of a bacterium? Something that would give me something like RefSeq’s “identical proteins” but for non-identical proteins? I think I’m really leaning into the title of the channel on this one
edit: I can make a pan-genome, but what I have is a list of gene names/locus tags for one strain, and I want to find the “equivalents” from other strains which aren’t identical
*Thread Reply:* closest thing i could think of immediately is a blast search and just exclude results that are >99% ANI? not sure if this is anything close to what you are looking for

*Thread Reply:* Hi! Something like this? https://www.microbiologyresearch.org/content/journal/mgen/10.1099/mgen.0.000499
*Thread Reply:* "(a) A fasta file containing a single representative sequence of each gene of the gene pool (https://doi.org/10.6084/m9.figshare.13270073, File F3)."
*Thread Reply:* I never came back to thank you both for your suggestions!! At the end some BLASTing and UniProt wound up working for me in this case, but Anvi’o seems to have a suite of tools that would help with this kind of question!

Hi All, I have a phd student who is looking into 16s rRNA analysis of oxalate samples, any recommendations on best aDNA protocol for those samples?
*Thread Reply:* Hey @s.wasef , If the data are not yet generated, it would be more interesting to look into metagenomics shotgun sequencing rather than 16s. They elaborate a bit on why in the latest review on aDNA https://www.nature.com/articles/s43586-020-00011-0.pdf
*Thread Reply:* Any suggestions on best extraction protocol to use with those samples? I did some sediment samples before, should I use same protocol?
*Thread Reply:* What kind of samples are these ?
*Thread Reply:* To follow on from Maxime: I think the 'original' paper was Ziesemer 2016(?) sci Rep. For the issue with 16S being too long for aDNA. Also possibly in Warinner 2017 Annual Reviews Human Genetics. @Tina Warinner?
I'm not sure I've heard of any aDNA protocols on that type of samples myself.
*Thread Reply:* Neither did I. We were brainstorming about best way to do that, and I thought to ask 😉
*Thread Reply:* Might just have to take a couple of boring Samples and try the standard EDTA extraction... I think a lot of people throw that at a lot of stuff and it sort of just always works
*Thread Reply:* (if it's safe)
*Thread Reply:* Yeah good idea! EDTA is always a good start.
non ancient DNA question - has anyone uploaded data to the ENA server and gotten this message - the project has been public for a week but no data is coming up 😕
No public data has been made available in this project yet. Awaiting submission and/or validation of data.
*Thread Reply:* Yes.
The Beethoven data has that at the moment (or last week when I was checking it)
*Thread Reply:* as a general rule ENA submission are a mess. sometimes it also takes a bit to show up. I would make sure the datasets show up under the right samples and project in the upload portal first, might be an association/upload issue and they could still be in the "waiting area"
*Thread Reply:* Ahh okay! That's good to know - thank you both! yep they look to be under the right project/sample and are linked (they even have a public status next to each file)
*Thread Reply:* You can also email their helpdesk to find out if something is stuck. My experience is that they normally quite helpful/friendly
*Thread Reply:* Yeah. I didn't say they were fast 🤣

Hello; I have a KRAKEN2 related question. I was trying to download taxonomy from NCBI to build a database using command "kraken2-build --download-taxonomy --db $database" but it came up with an error message "Downloading nucleotide gb accession to taxon map...rsync: [Receiver] failed to connect to ftp.ncbi.nlm.nih.gov (130.14.250.12): Connection timed out (110)". I cant figure out what this error is and how to resolve it? Looking online, they say that there is an issue with the script but I don't know how to modify the script and use it. Can someone help me?
*Thread Reply:* this is a super silly question: do you definitely have the ability to connect to the internet on your server? As a beginner, it looks like it's a connection problem rather than a code problem? Am I right in thinking this?
*Thread Reply:* Can be that NCBI has recently changed the path to ref genomes and Kraken2 hasn’t updated it yet. If you get this error for all ref genomes. It can also be that 1 (or more) particular genome changed its location or was dropped from the database
*Thread Reply:* Yeah thats the first thought I had. But we definitely have ability to connect to the server as wget works perfectly for downloading zip files from ncbi.
*Thread Reply:* To follow on the super silly question from Maria: for example I've seen people have this error when they've submitted the build command to their cluster, but only the head node has internet - worker nodes don't have access
*Thread Reply:* If you're using a cluster might be something to crack
*Thread Reply:* I am not using cluster either 😞
*Thread Reply:* Check the NCBI address Kraken2 attempts to connect to and try to wget a file from this address
*Thread Reply:* Thats what I working on now
*Thread Reply:* I also downloaded the old minikraken database, hoping that it has everything thta I need
*Thread Reply:* Hi @Kuldeep More,
It’s a known issue: https://github.com/DerrickWood/kraken2/issues/412
More or less, you have to go to the source code and replace the statements of ftp:// to https://. I faced the same issue recently and was afterwards to download everything.
 <a href="https://github.com/DerrickWood/kraken2">DerrickWood/kraken2</a>
<a href="https://github.com/DerrickWood/kraken2">DerrickWood/kraken2</a>
*Thread Reply:* Thanks Alex. I will try to use this and let you know.
*Thread Reply:* Okay now. I have successfully completed two steps for building a database. Kraken manual is not updated and hence its not that all helpful for beginners like me who don't play with the source code. So for people like me, I am posting what I did 😆
*Thread Reply:* To download taxonomy I used kraken2-build --download-taxonomy --db trialdb --use-ftp
*Thread Reply:* --use-ftp part is not in the Kraken2 manual
*Thread Reply:* now the next step is to dowload reference library. kraken2-build --download-library viral --db $DBNAME
*Thread Reply:* This command doesnt work because of the same ftp issue. So I dowloaded library directly from NCBI and then added that to the database.
*Thread Reply:* kraken2-build --add-to-library viral.1.1.genomic.fna --db trialdb
*Thread Reply:* And then building the database as in the manual
*Thread Reply:* Thanks for that! I just downloaded the database from https://benlangmead.github.io/aws-indexes/k2
I wish I had figured out your solution!

Hi everyone! Can someone guide me with NCBI Genome submission? Is it essential to create .ASN file or you can submit .fasta files?
Hello all, I have a HOPS-related question - In the original paper (https://doi.org/10.1186/s13059-019-1903-0), Hülner et al mention a HOPS database containing 6249 reference genomes, and state "HOPS database is available upon request." Does anyone know if this is accessible somewhere online (I'm thinking maybe it was made publicly available recently since), or do I still need to get in touch? Thank you for your help!
*Thread Reply:* Hi @Ophélie Lebrasseur, I have never sen that specific database but Ron Huebler replies emails and there are many people in this channel who might have it, or who built own Malt DBs with similar content. I built quite a few, they are not easy to share though because of their size. I would say ~6000 ref genomes is not a big DB and hence prone to low sensitivity / specificity of microbial discovery. The smallest Malt / HOPS DB that I have, comprises ~20 000 complete microbial refseq genomes. Please drop me a direct message if you would like me to try to share this (or other) Malt / HOPS DB with you. Alternatively, I can show you how to build your own, this requires a computer (node) with ~1 TB RAM though
*Thread Reply:* I guess HOPS / Malt DBs can be of general interest, right @James Fellows Yates? We recently published a few big KrakenUniq DBs and Bowtie2 indeces:
https://github.com/NBISweden/aMeta https://www.biorxiv.org/content/10.1101/2022.10.03.510579v1
So potentially I could try to share 2-3 Malt / HOPS DBs via FigShare if there is a general interest in the channel
*Thread Reply:* Hello @Nikolay Oskolkov, Thank you very much for the reply and for offering to share your database :) I think in a first instance, I'll try and get in touch with Ron Huebler - do you know if that is his email address: "huebler@shh.mpg.de" ?
Apart from that, I agree that HOPS/Malt db are commonly-used tools across the community and that maybe it would be worth investigating having a repository with these databases from papers maybe?
*Thread Reply:* No Ron is gone unfortunately and that email is probably dead. You should first try contacting Alexander Herbig. If he doesn't reply let me know, I think I know where the database is on our servers, I could see if I can put it on Zenodo or something
*Thread Reply:* I'm a bit wary of sharing malt databass though, there have been lots of problems with MALT recently and they are huge in most cases
*Thread Reply:* (just saying as other people have asked me the and thing in the past)
*Thread Reply:* Sorry to clarify: Ron does reply, so You could also try contacting Ron
*Thread Reply:* But I don't know if he ask access to his email nor the mpi-ev servers anymore
*Thread Reply:* And what Nikolay says (as usual)
*Thread Reply:* Great, thank you! I'll try to get in touch with both of them, and let you know!
*Thread Reply:* Ron replied my emails to his address huebler@shh.mpg.de, but it was ~2 years ago 🙂
*Thread Reply:* @James Fellows Yates Which tool would you recommend to replace MALT ? On top of the problems, it is also not handy at all to use
*Thread Reply:* all of them at once 🤣
*Thread Reply:* (which is why we've developed nf-core/taxprofiler: https://nf-co.re/taxprofiler#pipeline-summary)
*Thread Reply:* Or aMeta (😉) the approach I really like. You could probably swap out the HOPS bit with sam2lca after alignment with bwa etc and then damageProfiler (or a better stand alone tool that includes more stats)
Tbh the main obstacle is databases rather than the classification algorithm IMO. But the selection of the latter is driven by what you exactly are aiming to do.
*Thread Reply:* Database design I mean
Hello Everyone, back again with a basic question. Does anyone know how can I retrieve the list of refseq accession numbers based on the list of organism names (one refseq entry per organsim)? Chatgpt generated some code but it didnot work. If anyone here knows any tool, it will be really helpful. Thanks
*Thread Reply:* Maybe entrezcli or NCBI data?
*Thread Reply:* Tried both but not an option for this specific query. Its very easy the other way around
*Thread Reply:* Not at the top of my head without playing around with that
*Thread Reply:* @Maxime Borry might have an idea but I think he's travelling atm
*Thread Reply:* I will update here if it works
*Thread Reply:* Please do! I think taht would be really helpful
*Thread Reply:* If you're willing maybe you could even write a short blog post on teh problem and the solution for the <#C02D3DJP3MY|spaam-blog>? Then it would be more findable 🙂
*Thread Reply:* (outside of slack)(
*Thread Reply:* There: https://ftp.ncbi.nlm.nih.gov/genomes/refseq/assemblysummaryrefseq.txt
*Thread Reply:* what's the "some code" that didn't work from chatGPT ?
*Thread Reply:* @Maxime Borry Check this out
*Thread Reply:* something similar to what you want to do @Kuldeep More https://github.com/maxibor/ncbi-genome-download
*Thread Reply:* Thanks Maxime. This one requires the input file with taxid
*Thread Reply:* So the biomartr package works it seems. I tried only with 2 names, so that my R doesn't crash. Lets see if it works for 50
*Thread Reply:* ramp it up
*Thread Reply:* yaaaaay! it works
*Thread Reply:* So the package is available at https://cran.r-project.org/web/packages/biomartr/vignettes/Sequence_Retrieval.html#genomeset-retrieval
*Thread Reply:* In multiple retrievals I provided the list of organism names. Fantastic thing is, it automatically downloads only one refseq entry per organism
Hi all! I have a question for sedimentary and coprolite metagenomics people! Have you ever thought of comparing damage patterns in sediments/faeces with those in bone at a same locality? Are there differences in DNA taphonomy according to the substrate it is bound to? Has anyone published on this?
*Thread Reply:* I haven’t heard about anyone publishing this but maybe @irinavelsko knows.
*Thread Reply:* I haven't looked myself, but this is the paper that comes to mind where they might have https://www.nature.com/articles/s41598-018-28091-9
Hello everyone :)
I was wondering if anyone has recently (as in post-Brexit) had any experience transferring samples of CITES-listed specimens from the UK to the EU? My group needs to transfer samples from the UK to Sweden ideally by hand-carrying (rather than shipping it there). We cannot find definitive information of what paperwork is needed (other than the loan forms and certificates that the museum will give us), especially relative to airport customs both before the departure and upon arrival.
Thanks!
Hi all! When you're sequencing for publication and producing say 50-100 M reads for libraries, how much do you sequence controls? I've done screening with controls before, but my lab mostly does endogenous stuff so they'd never think of including controls on a deeper sequencing run. Would love your insight on this to handle controls properly 🙂
*Thread Reply:* We sequence controls to 5M reads, since they're usually pretty clonal. Deeper than that just amplifies more duplicates and doesn't add information
Hi everyone! There is this microbe that shows up a little bit too much in my results to be trusted to my taste 🧐: Pseudopropionibacterium propionicum (or Arachnia propionica). Is it only me or did you notice the same? Is there some explanation for it (better preservation or the like) ? Thanks 😊

*Thread Reply:* Hi Zoe, I had a look into some of my screening data, and I can find this bacteria in my sample too but not in a crazy amount (I'm working with animal material). It is an inhabitant of the animal and human flora so I'm not surprised. What do you mean by " a little bit too much"? Good luck🦠

*Thread Reply:* It is one of the most prevalent and abundant microbes in human dental calculus. It has been renamed multiple times, which may be why you’re having trouble tracking it. We wrote a bit about it in our 2017 review (see Figure 4):

*Thread Reply:* I think it's a highly diverse species/genus with insufficient genome diversity in databases. we also found it in chimps and gorillas (Fig 2), so it's probably a bog standard commensal (I suspect).
If you check for the number of multi-allelic positions when mapping you get a huge amount suggesting lots of strain diversity in samples too (Fig. S10)
So my feeling is that the single good genome attracts a lot of stuff making it seem stronger than it is
*Thread Reply:* Good point James - probably best to think of it as a group of related strains/species
*Thread Reply:* If you really have a lot of reads you could try de novo assembly
*Thread Reply:* Thanks @Louis L'Hôte @Tina Warinner and @James Fellows Yates for your answers 😊. It’s the microbe I find the most when screening samples so I thought there was something fishy about it. But what James suggests makes sense, perhaps the lack of a sufficient number of reference genomes for sister taxa in the database could explain why we so often end up finding this one. I checked the links and I think they show well that we don’t have enough genomes of this genus yet. Interesting 🧐!
*Thread Reply:* We might also discuss this a bit more somewhere in the PNAS paper, I can't remember now

Hi everyone! new to the community so good to meet you all! I've been working on some samples, and I get the following deamination pattern on a lot of my samples. I was wondering if anyone could help me interpret this. Thank you!

*Thread Reply:* I have seen that before, but I can't remember what caused it... what does your FastQC results look like? Could you maybe have some form of poly-A tails or something still in there?
*Thread Reply:* The FastQC results look good enough, there are some overrepresented sequences but they don't match the adapters used and no polyA tails.
*Thread Reply:* @Tina Warinner @Katerina Guschanski any ideas?
*Thread Reply:* Looks like there is some deamination on one side but not on the other side of the reads, which I assume might have something to do with the sequencing protocol (if this pattern is consistent across many samples as @Nicole Wagner mentioned), but I have no idea what it is 😞

*Thread Reply:* The same situation is mentioned in this paper. it may be due to the USER enzyme treatment after blunting. https://www.sciencedirect.com/science/article/pii/S0960982219307717#mmc4

*Thread Reply:* Cool, thanks @Yoshiki Wakiyama, do you mean this paragraph:
The specialty of the NEBNext Ultra DNA Library Prep Kit is that the adaptor provided by this kit contains a Uracil base that needs to be cleaved by the USER enzyme before the amplification step of the libraries. However, given that ancient DNA also contains Uracil bases especially at 5′ end, USER enzyme will also remove any Uracil residuals at 5′ end and thus resulted in the absence of deamination damage on 5′ end for the two samples.
*Thread Reply:* @Nicole Wagner did you use USER treatment?
*Thread Reply:* @Nikolay Oskolkov no, the initial illumina library prep was done with NEB Q5 hot start. However, after being sent to us, the illumina libraries were then converted into circular single strand libraries to be used with MGISEQ and i am not certain conversion kit they used.
*Thread Reply:* Ooooh that's really out of 'traditional' aDNA libraries, there coudl potentially be a lot of artefacts in there I imagine...
*Thread Reply:* @Åshild (Ash) (if you're availble() has worked with MGISeq before though
*Thread Reply:* (MGI and ancient DNA I mean...)
*Thread Reply:* Possibly related to @Yoshiki Wakiyama’s paper he posted above, maybe yo ucould contact the lab, they also did benchmarking: https://www.frontiersin.org/articles/10.3389/fgene.2021.745508/full
*Thread Reply:* (annoyingly without any plots...)
*Thread Reply:* (which I find slightly suspicious but who knows 😉 )
*Thread Reply:* Thank you! I will read through and follow up!
*Thread Reply:* An earlier BGI paper: https://academic.oup.com/gigascience/article/6/8/gix049/3888813
*Thread Reply:* (which looks quite similar to the MGI one funnkly enough)
*Thread Reply:* @Christian Carøe Was on the last paper, if he's also still around
*Thread Reply:* All the ancient DNA data that I’ve worked with that was generated by BGI was from double stranded libraries and behaved and looked like I would expect double stranded Illumina libraries to. I suspect it has something to do with the “circular single strand library” protocol you used, but I am unfamiliar with it
*Thread Reply:* I think I remember someone showing me these patters in single strand libraries… cannot it be something related to that?
*Thread Reply:* The jumpiness of the plot suggests there are very few reads going into the analysis. @Nicole Wagner Can you tell us how many reads are shown in this plot? If it is <1000, you can get strange and irreproducible patterns. Also, was a proofreading enzyme used as the first enzyme to amplify/index the library? If so, you could get artifacts like this that will suppress the damage pattern in ways that could be asymmetrical.
*Thread Reply:* Thank you for the info @Tina Warinner. There are about 4700 reads. There is no mention of a proofreading enzyme in the protocol I was given, but I will need to double check with the person who prepared the libraries.
*Thread Reply:* 4700 on-target reads should be sufficient in that regard (if evenly distributed across the genome), so I would lean more on polymerase or other artefacts of the library construction protocol
*Thread Reply:* @Nicole Wagner did you find the issue in the end?
*Thread Reply:* @James Fellows Yates we're pretty sure it's because they used the same prep technique as they use for normal metagenome sequencing. Fortunately, I'm going to the meeting in Tartu where i hope to learn more from experts in the field!
*Thread Reply:* What do you mean by that exactly? They used a modern kit or something?
*Thread Reply:* I'm not certain which kit they used just that they used the same kit they use for sequencing modern samples. I will learn more before the conference.
*Thread Reply:* Awesome! I would be curious to hear :)
*Thread Reply:* @Nicole Wagner do you mind if I copy and paste that image to a github issue? (sorry to bring up an old thread) it's so I don't forget to follow up in a few months if yo uwork out what the issue is
*Thread Reply:* ...wait actually
*Thread Reply:* Isn't it that with few reads?
*Thread Reply:* Of course, feel free to share! That is not mine, but looks very much like it.
*Thread Reply:* Aha! Then the Q5 is your culprit :D
*Thread Reply:* https://elifesciences.org/articles/73346
> This is in line with the DNA nucleotide misincorporation profiles expected for the type of DNA library constructed (Seguin-Orlando et al., 2015), which was caused by the Q5 polymerase being unable to read through 5' uracils, thereby excluding the typical 5' excess of C-to-T. MapDamage profiles were, thus, consistent with Cytosine deamination at 5'-overhanging ends as the most prominent postmortem DNA degradation reactions (Jónsson et al., 2013). I guess you have low number of reads which is why it's spikey
*Thread Reply:* More info: https://international.neb.com/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr
*Thread Reply:* This is the Q5 horse data downsampled to about 208 reads, it's smoother because the y-axis has more ticks and cut off is at 25bp not 10 in your plot, but I think it looks pretty similar

*Thread Reply:* Or even limited to 10bp:

*Thread Reply:* Thank you! this is extremely helpful. I will discuss this further with our sequencing team.
Hiya, does anyone know of any (or would be willing to share their) pre built database for krakenunique that would be suitable for sourcetracking? Soil, gut, skin, oral things included. Thanks :)
Dear all, I currently have a sample that contains ancient DNA for a specific bacterial pathogen (mean cov 4X). I also have a phylogeny built using modern reference genomes (based on concatenated core genes). What would be a suitable way of determining the closest modern lineage to my ancient pathogen DNA?
*Thread Reply:* Did you include your ancient genome in the phylogeny?
*Thread Reply:* If you haven’t done that you could extract a consensus of the core genes from the ancient genome if you reconstructed it by mapping, and use them to built the phylogeny
*Thread Reply:* I didn’t…. to generate the phylogeny I used a pipeline that uses as input the assembled draft genomes, but I couldn’t assemble a “good” genome from the ancient sample due to the low coverage
*Thread Reply:* ok, so you suggest me to map the ancient reads to the core genome I identified in the previous analysis, and get a consensus out of that, right?
*Thread Reply:* Although you may lose information at the beginning and end of the genes since it is likely that reads don’t map there
*Thread Reply:* Alternatively, you can use one of the modern genomes as reference to map your ancient reads and extract consensus for the genes in your core genome if it is easy for you to retrieve the coordinates of those
*Thread Reply:* that sounds good to me 🙂 I’m going to try to get that consensus sequence…. just one more thing 😊 what tool do you usually use to extract the consensus sequence from the mapping file?
*Thread Reply:* If you're using eager, you could use gatk unified genotyper + multivcfanalyzer. The latter will then produce fasta files - you can look at Aida's two stone age plague papers for examples
*Thread Reply:* And I think the eager tutorial for pathogens might have that
*Thread Reply:* oh that’s a great idea actually, and in Eager I can also input the coordinates that I want to focus the mapping on, right?
*Thread Reply:* https://nf-co.re/eager/2.4.7/usage#tutorial-pathogen-genomics---introduction
*Thread Reply:* Uhh can't remember about the coordinates... You can get basic coverage stats about gene features based on a bedfilw not sure about the mapping step :/
*Thread Reply:* You can use bedtools fasta to extract specific coordinates from a fasta. I can send a link to it later
*Thread Reply:* awesome, thank you very much @aidanva and @James Fellows Yates
*Thread Reply:* To clarify: I meant not sure you can do that within eager. Aida is right about bedtools manually!
Because it's a question that came back a few times here, here a updated indices for Kraken2 and KrakenUniq

Hi everyone. Quick question about the AMDirT - do we only enter shotgun sequenced samples or target-captured also? A paper came in with over hundred samples, most of them target-captured several times, but only a little 30 samples are shot gun sequenced according to the Supplement table. It's Clavel 2023: https://doi.org/10.1016/j.isci.2023.106787

*Thread Reply:* Depends on the table, if it's single genome then captured is OK.
Ultimately you want to include whatever was used in the genome reconstruction, in most cases just the targeted is what is used (as in most people don't mix shotgun and capture in their final reconstruction, I believe).
Does that help?
Hi all! Has anyone successfully gotten metaDMG to run from its conda installation? Would love to know if you bumped into any dependency/software/compilation issues and how you resolved them (currently getting an unspecified error that I can't seem to track down). Debating building it from scratch - has anyone had better luck with this?
*Thread Reply:* @Antonio Fernandez-Guerra one for you methinks 😁
*Thread Reply:* (More info: I'm running this on a remote linux server where I don't have admin permissions)
*Thread Reply:* I think that the core metaDMG-cpp file doesn't exit in the conda environment given by the metaDMG-core git page, so I've been wrangling with a separate installation of it from the metaDMG-cpp git page (to no success yet), but let me know if I've just missed it somehow!

*Thread Reply:* I had been having hard time with it as well. But this worked for me: https://github.com/metaDMG-dev/metaDMG-core/issues/11#issuecomment-1546828093
<a href="https://github.com/metaDMG-dev/metaDMG-core">metaDMG-dev/metaDMG-core</a>
*Thread Reply:* Gotcha! Thanks for the info. I stumbled across that chunk of code and got metaDMG-core installed (and presumably) running. Did you have to download and compile metaDMG-cpp separately?

*Thread Reply:* Hi Carly, I installed and updated metaDMG some while ago and here are the steps I used. Try if that helps! To answer your questions: I installed metaDMG-cpp separately on the server. During the latest update I needed to specify the version of logger_tt to 1.7.2 (see script below) to make it work. ```# install updated metaDMG with dependency requirements mamba create -n metaDMG2 python=3.9 conda activate metaDMG2
mamba install -c conda-forge libdeflate=1.6 mamba install -c bioconda htslib=1.10 conda install -c omnia eigen3
mamba install -c bioconda -c conda-forge htslib eigen cxx-compiler gsl pip install git+https://github.com/metaDMG-dev/metaDMG-core@stopiferrors_branch pip install iminuit numpyro joblib numba logger_tt==1.7.2 psutil pip install metaDMG[all]
now install metaDMG-cpp
git clone https://github.com/metaDMG-dev/metaDMG-cpp.git cd metaDMG-cpp make
update metaDMG-cpp
conda activate metaDMG2 cd ~/metaDMG-cpp make clean git pull https://github.com/metaDMG-dev/metaDMG-cpp.git make```
*Thread Reply:* Thanks for the info - this worked but metaDMG-cpp won't compile 😕 I'm thinking the issue is a conflict with my cluster's default environment (gsl/gcc) so hopefully our tech support can help resolve the problem.
Hello, if you're submitting samples to the ENA where you've got multiple species' genomes to submit from one sample (they're from host-associated metagenome), do you put the host species ID in the tax_id section of the sample checklist? I also don't know what collection date to put down - do people tend to put date of excavation/ date of sampling or date the sample actually came from? I may also be using the wrong checklist for submitting samples - currently I've got the minimal ENA default sample checklist, but if anyone thinks I should use a different one please let me know! Thanks! 😄
*Thread Reply:* There should be two taxids field I think, one for host and the other for the sequences themselves. I believe there is a dedicated tax I'd for metagenome
*Thread Reply:* second stupid q- do people put excavation date for collection date or the actual date associated with a sample? or something else (like date the material was actually sampled for DNA analysis?) Thanks 🙂
*Thread Reply:* The very latter is my understanding
*Thread Reply:* thank you! sorry for all the questions, just don't want to submit the wrong thing 😅
*Thread Reply:* IT's likely form here: https://genomicsstandardsconsortium.github.io/mixs/0000011/
*Thread Reply:* So sampling for DNA I think
another (potentially) stupid question: how much difference does it make to use a sequencing strategy of e.g. PE50 compared to PE150? I know my target sequences are short (~50 bp), so from a technical point of view, PE50 is the way to go. However, from a financial point, the same number of reads costs twice as much when using PE50 (compared to PE150). If I understand the theory behind it, PE150 will generate more overlapping and uninformative reads so I would need a larger number to get the same amount of informative output, but I wonder how big that effect is..
*Thread Reply:* No stupid questions! The number of reads generated depends on the flow cell, not the sequencing chemistry, i.e. a library sequenced on a flow cell at 2x50 will have the same number of demultiplexed reads as a library sequenced on the same flow cell at 2x150. The metric you might be seeing increase with increasing sequencing length is Gb/Tb of output, which just refers to the number of bases sequenced, i.e. a lib sequenced at 2x50 will have fewer sequenced bases than a lib sequenced at 2x150 but the same number of reads. Because of this a 2x50 run is generally cheaper than a 2x150 run, but cost also depends on where you get your sequencing done and which flow cells the centre uses the most. Some centers don’t even offer 2x50 chemistry.
*Thread Reply:* If your inserts are only 50bp long, 2x50 is plenty if you can find it cheaper than 2x150 lanes. You should prioritize finding the flow cell that will produce the most reads (millions or billions these days) for your budget, not necessarily the most output (Gb, Tb)
Hi everyone! I don't really know where to ask this so don't hesitate to redirect me somewhere more adequate. @Emrah Kırdök and me had to move servers recently and we encounter this error when running MALT. We think it is linked to memory somehow but the job doesn't seem to use up all the memory available on the node either. Here is the example for a job. Log file: ```Version MALT (version 0.6.1, built 25 Oct 2022) Author(s) Daniel H. Huson Copyright (C) 2022 Daniel H. Huson. This program comes with ABSOLUTELY NO WARRANTY. --- LOADING ---: Reading file: results/MALTDB/maltDB.dat/ref.idx 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (0.7s) Reading file: results/MALTDB/maltDB.dat/ref.db 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (573.5s) Number of sequences: 2,165,573 Number of letters: 47,192,673,378 LOADING table (0) ... Reading file: results/MALTDB/maltDB.dat/index0.idx Reference sequence type: DNA 100% (0.0s) Reading file: results/MALTDB/maltDB.dat/table0.idx 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (60.1s) Reading file: results/MALTDB/maltDB.dat/table0.db 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (1,344.0s) Table size: 46,493,548,843 Loading ncbi.map: 2,396,736 Loading ncbi.tre: 2,396,740 Reading file: results/MALTDB/maltDB.dat/taxonomy.idx 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (0.2s) --- ALIGNING ---: +++++ Aligning file: results/CUTADAPTADAPTERTRIMMING/ldo058-b1e1l1p1withouthuman.trimmed.fastq.gz Starting file: results/MALT/ldo058-b1e1l1p1withouthuman.trimmed.rma6 #
A fatal error has been detected by the Java Runtime Environment:
#
SIGBUS (0x7) at pc=0x00007f4e7399d199, pid=70370, tid=70691
#
JRE version: OpenJDK Runtime Environment (18.0.2.1+1) (build 18.0.2.1+1)
Java VM: OpenJDK 64-Bit Server VM (18.0.2.1+1, mixed mode, tiered, compressed class ptrs, g1 gc, linux-amd64)
Problematic frame:
V [libjvm.so+0x813199] HeapRegionManager::par_iterate(HeapRegionClosure, HeapRegionClaimer, unsigned int) const+0x99
#
Core dump will be written. Default location: /cfs/klemming/projects/snic/ancient_microbes/pochonz/aMeta/core
#
An error report file with more information is saved as:
/cfs/klemming/projects/snic/ancientmicrobes/pochonz/aMeta/hserr_pid70370.log```
It's not very explicit. A binary "core" file is generated and a very long "hserrpid70370" (attached). And in the slurm log it just notifies that there is an error by adding "/usr/bin/bash: line 1: 70370 Aborted" before the malt command and the usual
unset DISPLAY; malt-run -at SemiGlobal -m BlastN -i results/CUTADAPT_ADAPTER_TRIMMING/ldo058-b1e1l1p1_without_human.trimmed.fastq.gz -o results/MALT/ldo058-b1e1l1p1_without_human.trimmed.rma6 -a results/MALT/ldo058-b1e1l1p1_without_human.trimmed.sam -t 128 -d results/MALT_DB/maltDB.dat -sup 1 -mq 100 -top 1 -mpi 90.0 -id 90.0 -v &> logs/MALT/ldo058-b1e1l1p1_without_human.log
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
sacct just says "FAILED" and seff shows that it was far from using up all the memory of the node:
Job ID: 1849320
Cluster: dardel
User/Group: pochonz/pochonz
State: FAILED (exit code 1)
Nodes: 1
Cores per node: 256
CPU Utilized: 00:49:19
CPU Efficiency: 0.55% of 6-05:45:36 core-walltime
Job Wall-clock time: 00:35:06
Memory Utilized: 418.67 GB
Memory Efficiency: 23.39% of 1.75 TB
I don't think it is an error in the code because many malt jobs work and other don't. But it's not stochastic because the ones that don't work, won't work when just rerunning. We think it might be that the jobs are not allowed to use up all of the node RAM as memory somehow, but we are not sure if there could be some magic piece of code to prevent this from happening. The server support is not supportive at all, so if it is something that needs to be done on their end, we might as well have to move again 😅
Anyone familiar with that error and knows why it happens and maybe even how to fix it?
*Thread Reply:* > We think it might be that the jobs are not allowed to use up all of the node RAM as memory somehow, This might be the error because f**ck Java
*Thread Reply:* Because it does half second peak memory requests
*Thread Reply:* And the memory loading is stochastic...
*Thread Reply:* But I don't know any more than that 😞
*Thread Reply:* Yeah I hate java too 😂. Thanks anyway 😊
*Thread Reply:* Sorry it's not more helpful... but it sounds very similar to what we have on our cluster that uses a Java-hating schuldar
*Thread Reply:* I guess if I don't find a fix, I'll have to splice the fastq files in two parts, until we find a new cluster
*Thread Reply:* I was having the same issue with java with another software. For me the problem was that in our cluster managed by slurm they set a java memory someplace I could not access to and could not change not even with the -Xmx flag. But if you haven’t tried yet, and if you have it set up differently in your cluster, maybe you can increase the memory from the default 64m to -Xmx1024m maybe…(sorry very naively answering to your question).
*Thread Reply:* Hey @Maria Lopopolo, this is a relevant suggestion and sorry that I forgot to mention that I had already adapted the heap space. Thanks anyway 😊

*Thread Reply:* Hi,
Did you change the Xmx value ?
Because in the log file you attached, it says -Xmx2000G which is a request of 2TB. However, according to the seff output you have only 1.75TB. Also, in the log file, it says that you program tried to access memory address that are not existing.
So maybe one solution would be to reduce the request of memory that java is doing (to less than 1.75TB because the system need also memory to manage other processes).
*Thread Reply:* Hey Julien, I think I already tried before with -Xmx1000G but I can try again with -Xmx1500G and see what happens. Thanks 😊
*Thread Reply:* I had been told by a colleague that it was no problem to make java believe that it had more memory available than in the truth. He might have been wrong there 😅
*Thread Reply:* I don't know, but you clearly have a memory issue as it says your process made a core dump.

Hi All,
Is there anyone who works with the CARD (comprehensive antibiotic resistance database) database? I’ve aligned it the reads to CARD v.3.2.7 with MALT and now I am trying to open the .rma6 file in MEGAN but I am having trouble. The CARD (C) button on the tool bar vanished in the newest MEGAN version, and even with the older MEGAN v.6.23.4 the CARD button doesn’t work. • Could this be a problem with CARD being recently updated (June 2023) and MEGAN do not have an updated mapping file? • Could this be a problem with how the alignment is done with MALT? Any insights are much appreciated! Thank you!
*Thread Reply:* Hi Christine, I worked with the CARD db, I used Blast though and not MALT so I cannot help you with that. I made a Blast DB from Card and run the analysis as described in the study by Brealey et al, 2020 (https://academic.oup.com/mbe/article/37/10/3003/5848415). I hope this helps!
*Thread Reply:* This helps, thank you Claudio!
Hi all, I need to send a pool of sedaDNA libraries from the Netherlands to Germany. The heatwave got me thinking about what would be the best way to secure the DNA quality. I was thinking of a box filled with dry ice (maybe not necessary) or ice packs. What would be your suggestions? Thanks already!
*Thread Reply:* I would send DNA (extracts, libraries etc.) always on dry ice, regardless of a heatwave.

Hello everyone! I am conducting an analysis on an ancient bacterial genome, and I obtained these graphs using mapDamage. I would like to know if any of you know how to interpret this kind of results. Thank you 🙂

*Thread Reply:* I think they look OK (although a bit low), the random bump could be maybe somelike like the end of the adapter still there or something like that?
*Thread Reply:* Checking FastQC results might make sense to doublec hcek
*Thread Reply:* I also thought that the bump at the end might be due to some weird pattern / low-quality bases left. Regarding whether the deamination looks convincing, I would say I doubt so since the frequency of C/T polymorphisms does not exceed (or very marginally) the frequencies of all other polymorphisms (the grey lines). This however may mean that together with truly ancient reads, there is a bunch of mis-aligned modern reads, so the quality of alignments should be checked
*Thread Reply:* Its low but apart from the bump, it looks ok.
*Thread Reply:* Congrats on the ancient(ish) genome @Luisa Sacristan! 😬
*Thread Reply:* @Luisa Sacristan if you work out what it is, please let us know, then we can add it to the little book of smiley plots 😄
*Thread Reply:* https://www.spaam-community.org/little-book-of-smiley-plots/
*Thread Reply:* Thank you all! I'll check based on your suggestions.
*Thread Reply:* @Luisa Sacristan do you mind if I copy just the image to the github repo for the little book of smiley plots?
*Thread Reply:* Just wondering: besides the weird bump on the 5' end, isn't anyone suspicious about the non-G-A grey substitution on the 3' end that is higher in frequency than the G-A substitution? To me that would be a red flag in terms of accepting the reads from this organism as showing damage patterns. It seems like some other processes are taking place: Mixture or organisms, mixture of damaged and non-damaged reads, ligation biases during library prep, etc.
*Thread Reply:* The grey lines are also elevated 5p, but in both cases the grey 'artefact' is consistent -ish. which is why I think it's a lab artefact at least. If it was organism related everything would be elevated.
*Thread Reply:* (also interesting it's one grey line on 3p, and not 5p)
*Thread Reply:* Exactly, it's the 3' that would worry me more than the 5'
*Thread Reply:* The depurination pattern is also odd. A should be enriched, not depleted at 5’ -1. @Luisa Sacristan How was the library built? This looks like an enzyme issue to me. Was this built with Nextera or another similar commercial kit?
*Thread Reply:* @James Fellows Yates It's ok 🙂 you can copy the image
*Thread Reply:* @Tina Warinner yes, the A behavior is not normal. I'm waiting the laboratory answer about the lib preparation.
*Thread Reply:* Was there anyhting in your FastQC resutls/
*Thread Reply:* @James Fellows Yates there were no adapter content and Per base sequence quality was good.
*Thread Reply:* @James Fellows Yates

*Thread Reply:* Then it maybe a ligation bias or something like Katja said
Hi everyone! quick question…. for programs like MapDamage, is the reference used required to be a complete genome, or is it possible to also analyse your reads with a draft genome available as separate contigs?
*Thread Reply:* Contigs are fine!
*Thread Reply:* @thanks Nikolay, but I guess somewhere in the analysis I’ve got to set a different parameter/setting in comparison with the case a full reference is used?
*Thread Reply:* I’m asking because I got an “odd” pattern in my plot, let me show you…
*Thread Reply:* This is what I got when using a complete reference

*Thread Reply:* and I got this when I used a fragmented reference genome

*Thread Reply:* Mmmm, no, the reference you provide with -r flag can be any level of assembly. It just needs to match the reference used for producing the bam-alignment
*Thread Reply:* To me, both look similar
*Thread Reply:* mmm but why do I get all those extra points in the second one and not in the first one?
*Thread Reply:* You mean the grey lines?
*Thread Reply:* no, I mean in the nucleotide frequency plots (the top 4 panels)
*Thread Reply:* Hmm, not sure, let me think a bit what exactly and how is being plotted in the top 4 panels
*Thread Reply:* Intuitively, non-optimal assembly should result in more mis-alignments and more noisy mean allele frequencies. But what exactly all those scattered points mean, I do not know right now
*Thread Reply:* @Alex Hübner?
*Thread Reply:* (also @Guillermo Rangel this might be one for: http://www.spaam-community.org/little-book-of-smiley-plots/)
*Thread Reply:* @Guillermo Rangel Do you have many contigs in the fragmented reference? How many?
*Thread Reply:* I suspect that one point might be one contig
*Thread Reply:* the assembly has 264 contigs
*Thread Reply:* it’d make sense if each point represents a different contig
*Thread Reply:* and sure @James Fellows Yates , I’m happy to contribute a figure to the book 🙂
*Thread Reply:* Shout when you know the cause!
*Thread Reply:* @Guillermo Rangel My interpretation is that in the case of complete genome you have one (or a few very similar) reference, therefore one point or a few very close points, while with the fragmented genome you have a lot of heterogeneity in terms of mapping to each contig. If you go to the raw output from mapDamage, there should be 264 points for the top 4 panels, i.e. one of the dimensions of the matrix used for constructing the plots in the top 4 panels should be 264
*Thread Reply:* hey @Nikolay Oskolkov I think you’re right 🙂 I just checked the files and manual… so for the misincorporation plot the program combines the results for all the reference sequences used (in my case all the contigs), but for the dnacomp plot it doesn’t, so I think each point per position represents a different reference sequence
*Thread Reply:* So the wierd pattern is alignment against a single contig?
*Thread Reply:* I think that for each position in the dnacomp plots each point represents the nucleotide frequency for each one of the contigs in the target assembly, which in my case had 264 contigs
*Thread Reply:* and I guess the connected dots are the mean or the median for each position
*Thread Reply:* @Guillermo Rangel if you're happy with GitHub, you can make a PR:
https://github.com/SPAAM-community/intro-to-ancient-metagenomics-book/
(instructions are on teh rEADME, but you can probably copy from the other pages 🙂
*Thread Reply:* IF you're not, please could you send me the following mapDamage/DamageProfiler files:

*Thread Reply:* And a short descriptio nof what causes the misincorproation/smiley plots :)
*Thread Reply:* Hi @James Fellows Yates I’m going to try to send a PR, but where in the repo should I place the files? Do you want me to send you only the .txt files you requested?
*Thread Reply:* There are instructions on the rEADME 🙂
*Thread Reply:* Oh, I think I’d cloned the wrong repo… https://github.com/SPAAM-community/intro-to-ancient-metagenomics-book/
*Thread Reply:* ok, I’ll change it and follow the correct guidelines
*Thread Reply:* Did I put the wrong link
*Thread Reply:* yeah you sent me the one for the intro to metagenomics book, but it should be fine now 🙂
*Thread Reply:* OOOPS my bad
*Thread Reply:* too many book ideas 😅
*Thread Reply:* James, my data was generated with mapdamage instead of damage profiler, is that still fine?
*Thread Reply:* The output data should be identical
*Thread Reply:* cool, ok I think I’m almost done, I’m going to push changes shortly
*Thread Reply:* Look forward for the PR!
*Thread Reply:* cool… in order to push changes I should fork the repo right? just realised I don’t have permissions to push directly 😅
*Thread Reply:* Either I add you a collaborator and you can push to a branch, or you can do a fork
*Thread Reply:* I’ve forked the repo 🙂
*Thread Reply:* @Guillermo Rangel could you add a short description to the end of the .qmd file of what causes the damage profile?
*Thread Reply:* just did 🙂
*Thread Reply:* one question though…. the figure in the book will only display the damage plot, or also the dna composition plot?
*Thread Reply:* Technically just the damage plot, but we can include more images below the text if you can recreate the plot!
*Thread Reply:* Ok I realise now that in your specific case we will actually need the bigger file 🤦
*Thread Reply:* (with all mutations)
*Thread Reply:* (the misincorporation table - my bad @Guillermo Rangel!)
Hi all! Back again with another metaDMG question - is there an easy way to run it on repaired libraries (mine are UDG-half)? Does it account for this already?.... if not, where would I edit the algorithm by hand?
*Thread Reply:* (though I feel like UDG-half libs already follow the expected pattern somewhat, just in an extreme sense, so maybe will just lower the significance threshold I set)
*Thread Reply:* Actually.... after more exploration I don't think it matters and metaDMG is definitely accurately sorting ancient vs. non ancient reads

Hi all. Can someone recommend a good review paper or a nice resource to learn about the state of the art/best practices of paleo-proteomics?
*Thread Reply:* https://pubmed.ncbi.nlm.nih.gov/29581591/
or the mega (40page!)
https://pubs.acs.org/doi/10.1021/acs.chemrev.1c00703
*Thread Reply:* this one too can help: https://www.science.org/doi/epdf/10.1126/sciadv.abb9314
*Thread Reply:* Unfortunately, the "guide to ancient protein studies" does not allow access for me (my institution). Does anyone have access and could share it with me?
*Thread Reply:* You can try

*Thread Reply:* Press the Univ. Turin button
*Thread Reply:* And then the bottom 😉

*Thread Reply:* But in the mean time (on behalf of @Christina Warinner)

*Thread Reply:* Sneaky 😉 @James Fellows Yates Thank you so much for forwarding the article! this helps a lot @Christina Warinner


Hi everyone. I was curious if anyone had ever tried using a kit such as this to increase the proportion of endogenous DNA to contaminant DNA in a sample? My thinking was that the sample could be run through the column in the supplied buffer, binding fragments above ~150-300 and letting the shorter endogenous DNA through, which could then be extracted from the buffer using something that recovers shorter fragments.
*Thread Reply:* I think the group of Matthias Meyer at MPI EVA has been using electrophoresis gels for size selection. For this, they ran the library samples on a gel and then cut out the part of the gel they want to keep. However, I am not sure if there is a particular protocol for this that they published. You should just keep in mind that you loose a lot of DNA in these processes so you need plenty to start from.

Hello, I am not sure if this is a question for this channel or for nf-core/eager slack but I had a question about AdapterRemoval, I was wondering when it was appropriate to use --preserve5p ? I am working on shotgun dsDNA library-prep sequences. I am trying to select the parameters for AdapterRemoval and am having trouble figuring out what kind of parameters I should think about with my sequences. If anyone has any suggestions on how I can approach this I would greatly appreciate it. Thank you!

*Thread Reply:* The reason why this was added was in the case of very low coverage data which had a very high duplication factor.
The problem was when trimming was carried out, in some cases of a set of duplicate reads, when doing quality trimming some would be trimmed at 5p but others not. Which led to the deupping tool too incorrect (baby waking up, continue Kater)
*Thread Reply:* ...Incorrectly not dedup some of the duplicates because there 5p was trimmed more than others
*Thread Reply:* So essentially you left in duplicates, which artificially inflated your coverage and cause false positives in terms of confident variant calling
*Thread Reply:* So: if you have low coverage data, with a high duplication rate, then it maybe recommended
*Thread Reply:* Ohh that makes sense! Thank you so much for your help and for connecting me to the correct eager channel!
Hey all! Lab related question. I'm using a single stranded protocol (Gansauge & Meyer, 2019), and for the ligation of the first adapter they use PEG-8000. We've found that we can only order it in a kit and keep ending up with a lot of other unused reagents. I was wondering if anyone has ever compared PEG-6000 or PEG-4000 vs PEG-8000 for ancient libraries? I used some PEG-4000 for captured samples and that seemed the have worked fine, but trying to figure out what to do for some other samples for shotgun sequencing.
*Thread Reply:* I asked in our departmant chat and got the following response
Hey James, related to your PEG question - Marie and Matthias have heavily optimised the ssDNA library prep, so I am sure they tested various PEG concentrations (especially since the higher molecular ones are super viscous and pain in the ass to work with). Matthias is currently away until some time next week - but he would be the best person to ask and he's quite responsive to these types of technical questions 🙂 So my recommendation is that the person just shoots an e-mail to him and will get the best advice 😉
*Thread Reply:* I usually order PEG8000 as a powder and prepare my own aliquots, in case that is an option for you
*Thread Reply:* Thanks both! I will send Matthias an email, and look into the powder 🙂

*Thread Reply:* Another response!

Hi all! We have a similar issue to Meriam's. We use the Roche extender columns for the Rohland etal 2018 protocol, however most of the buffers from the Viral Kit get piled up, unused. Has anyone found an alternative to buying the whole kit? As far as I know the extenders are not sold separately.
*Thread Reply:* Exactly the same for us, no solution but also keen to hear other ideas
*Thread Reply:* Same here! I haven’t found the columns sold separately

*Thread Reply:* I’ve seen a protocol that was published recently for soil and sediment samples and there they use these EconoSpin® DNA Only Maxi Spin Columns. https://epochlifescience.com/products/econospin%C2%AE-dna-only-maxi-spin-column?variant=44335099412771

*Thread Reply:* And this is the protocol that was published: https://www.protocols.io/view/inhibitor-free-dna-extraction-from-soil-and-sedime-bp2l6957zlqe/v1

*Thread Reply:* I don't think we use the viral Kits either:

*Thread Reply:* https://www.protocols.io/view/illumina-double-stranded-dna-dual-indexing-for-anc-4r3l287x3l1y/v2/materials
*Thread Reply:* Thank you @Anna Chagas I'll check it out! @James Fellows Yates it's the DNA extraction protocol, forgot to mention 😁
*Thread Reply:* Ah ok... sorry, I thought Rohland 2018 was the UDG half 🤦
Hi all! We’ve just switched clusters and I’m trying to run MALT/HOPS and troubleshoot. Seems like on some file sets it’s working fine and sometimes it throws INFO: java.util.concurrent.ExecutionException: java.lang.NullPointerException — this may not be a MALT/HOPS issue per se but if anyone has seen it before I’d be very grateful!
*Thread Reply:* Does this pop up when you’re running malt run or build?
*Thread Reply:* You'll need to provide more information around the error too - that error is the most generic of all Java errors possible...
*Thread Reply:* Ah, heck. Okay, it seems like at least in my most recent run, the first error that seems to lead to a cascade of disasters is a “INFO: Danger empty keys in File” at the MaltExtract step. My problem seems very similar to this one. https://github.com/rhuebler/MaltExtract/issues/2
*Thread Reply:* So I suppose I could just hop on the github but was wondering if anyone else has dealt with this!
*Thread Reply:* Can you open your RMA6 file in Megan?
*Thread Reply:* Empty keys in file would suggest to me it's possibly a broken rma6
*Thread Reply:* But otherwise given the repo is ignored it seems, you should email Alexander Herbig
*Thread Reply:* Ooh, will check up on the RMA6 files and email Alexander if need be. Thanks so much James!
*Thread Reply:* Aha, something has indeed gone awry with the RMA6. MALT output says out of 32 M reads, ~900K were aligned; MEGAN shows the RMA6 file as having 900K reads but 0 assignments. Thanks James, will send this info to Alexander and hope for the best!
*Thread Reply:* Glad you at least solved the first bit!
Is anyone interested in reviewing this paper for PeerJ? You should be able to sign up using the link below.
Article Id 88906 <https://nam10.safelinks.protection.outlook.com/?url=https%3A%2F%2Fpeerj.com%2Fsubmissions%2F88906%2F&data=05%7C01%7Csvw5689%40psu.edu%7Cec3de90afdf84024dafd08dba961df11%7C7cf48d453ddb4389a9c1c115526eb52e%7C0%7C0%7C638290010050724252%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000%7C%7C%7C&sdata=2YS91HQ1jtRAJN0fJllTkF7R%2FwFkdDvIL2Am6Fw26PM%3D&reserved=0>
Title
The relationship between leptin and periodontitis: a literature review
Abstract
Leptin is a peptide hormone that regulates energy balance, immune inflammatory response, and bone metabolism. On the other hand, periodontitis is a local inflammatory disease that progressively weakens periodontal support tissue, eventually leading to tooth loss. Several studies have demonstrated a relationship between leptin and periodontitis. This article reviews the existing literature and discusses leptin’s basic characteristics, its relationship with periodontitis, and its effects on periodontal tissue metabolism.

Hi all, for shotgun sequencing sedaDNA, how do you decide how many reads to target per sample? I’m trying to put together a budget and there seems to be a big range in the literature. Thank you!
*Thread Reply:* Depends on that you want to do!
*Thread Reply:* Can you provide more info on the goal of the project?
*Thread Reply:* Ha it always does right? The goal of the project is to understand timing of human arrival and associated environmental change through the late pleistocene and early holocene in currently submerged subarctic coastal sites. We will mainly be metabarcoding core samples to look at plant and animal community change as well as signatures of sea level rise (i.e. shift from terrestrial plants to algae), but my collaborators are interested in doing some shotgun sequencing as well and we’re just trying to figure out how much sequencing to budget for (approximately)
*Thread Reply:* I see. I meant more technically - what do you analytically want to do? Just taxonomic profiling? de novo assembly? functional profiling?
*Thread Reply:* But maybe we can crowd source some response (and if you're willing!) maybe yo ucould write a short blog post? @Kevin Daly did this for one of his questions for pathogen screening.
https://www.spaam-community.org/blog/2023/04/07/blog-numreads/
A companion piece for sedaDNA would be really nice 🙂
*Thread Reply:* To crowd source ideas, off the top of my head:
@Pete Heintzman @Antonio Fernandez-Guerra @Anan Ibrahim @Kadir Toykan Özdoğan @Linda Armbrecht @Barbara @Benjamin Vernot @Chenyu Jin (Amend) @Eric Capo @Jamie Wood @Kevin Nota @Laura Epp @Merlin Szymanski @Mike Martin @valentinav
Maybe they could chime in?
*Thread Reply:* Just to give one individual example, don't know if it helps. We tried 8-16-32 million reads per sample for some samples from the same site for screening. What we noticed was that we lost a lot of the diversity with 8 million reads but there was not much difference in that sense between 16 and 32. But it is just one example and just for screening to see what is there, not for any deep analysis.
*Thread Reply:* What sort of screening/analysis?
*Thread Reply:* To see which types of plants and animals are present in family level
*Thread Reply:* (sorry I mean which technique: taxonomic profiling? Mapping? De novo?)
*Thread Reply:* taxonomic profiling by using bowtie2 for alignments

*Thread Reply:* I agree with Kadir. It really depends on the type of sample, and how rich your sample is. When we do screening for the preservation of DNA, and have a superficial idea of which plant species were present. We tend to aim between 2-5 million reads and do a mapping approach with an LCA. That usually gives enough data to get some idea about the plants based on nuclear DNA and to see if there is DNA preservation. If we want to get a bit more resolution and have more chance you will pick up an animal signal (in archaeological sediments or lakes), we aim for at least 20-25 million again with a mapping approach with an LCA. Again it will really depend on your samples and how much you will get. In the end, it is a question of which sequencing platform you will use NovaSeq is cheaper per read than HiSeq, and the number of samples you have.
*Thread Reply:* Thank you for these responses! This is super helpful. We are planning to do taxonomic profiling, but I think my collaborators are not super clear on what specifically they’re hoping to learn from the shotgun data (this is a sedaDNA add on to a larger project), so this info should help us nail down the details. Thanks so much!

*Thread Reply:* Also as Kevin mention the prices for secuencing are important, depens of how many samples do you have and how deep do you want to go in. But for taxonomic profiling 5 million reads, cold be enough! Good luck!

Hello dear community! Hope you are all fine. I have one question about the analysis of coprolite (very old one), and I hope someone here has more experience than me with these kind of data. Basically, there are a couple of coprolites which have been sequenced using dsDNA library-prep approach. I was wondering if there was a bit of human DNA inside thus I applied both CoproID and nf-core/eager on the same samples to compare the results. I get very discordant results between the two but I am not understanding exactly why this is happening. I share you here an example from one sample: From CoproID I obtain a good prediction as being human. I attach both the summary picture than the deamination profile obtained. Then When I run eager I obtained a completely different result with almost no deamination at all in the same sample. I have used the same reference for both (GRCh37) and default params for both analysis. Does anyone can help me understanding what is going on there? What am I missing? Thank you people!



*Thread Reply:* coproID damage plots are only from the damage filtered reads, so that might explain why you get this difference. How many reads do you have aligned to Humans with coproID ?
*Thread Reply:* Hello Maxime, thank you for the explaination. I get 72303 aligned on human, 1870 aligned on dog and 2850 ancient human, 87 ancient on dog.
*Thread Reply:* So you think the problem is that on eager the damage plot is done on all human not just the ancient part, with respect to what CoproID does? is it right?
*Thread Reply:* Given what Maxime says, that makes sense to me. I'm not really sure why you would do a damage plot only showing damaged reads, though?
*Thread Reply:* Seems like that runs the risk of masking quite a bit of contamination?
*Thread Reply:* that's what i suspect @Andrea quagliariello
*Thread Reply:* a damage plot for a qualitative and visual confirmation of the damage
*Thread Reply:* Ok Thank you @Maxime Borry. So the damage plot is draw on the output of pmdtools? because I do not find the same number of ancient reads in the bam of pmd output with respect to the number of ancient aligned presented in the final csv (coproID_result).
*Thread Reply:* So to summarize: coproID is just there to help you pinpoint the source/host of your coprolite. The rest of the classic aDNA authentification steps have to be done with your classic aDNA pipeline (nf-core/eager, or else)
Hello everyone, does anyone have a nice package or script to make circos plots to compare assemblies? They don’t seem easy! Any suggestion appreciated! Cheers!
*Thread Reply:* Hi Maria,this one is not too bad https://github.com/metagenlab/mummer2circos
*Thread Reply:* I've not used them myself but if I you are a fan of R, I've heard of:
https://r-graph-gallery.com/224-basic-circular-plot.html https://cran.r-project.org/web/packages/BioCircos/vignettes/BioCircos.html https://cran.r-project.org/web/packages/RCircos/vignettes/Using_RCircos.pdf
*Thread Reply:* but AFAIkj, CIRCOS is never easy)
*Thread Reply:* Also: https://moshi4.github.io/pyCirclize/
*Thread Reply:* Thank you so much for the suggestions! I will have a look at all of them!
*Thread Reply:* also do you do recommend annotating them all with the same tool beforehand?
*Thread Reply:* Yes ! Different tools will give you different results, so be careful 😉
*Thread Reply:* Also, if you have only 2 genomes to compare, I would definitely recommend dotplots (this one is nice and interactive: https://dgenies.toulouse.inra.fr/)
*Thread Reply:* I have like 5 but also can do a closer inspection with 2.
*Thread Reply:* and if they're too divergent for circos like plot, you can give a try to pangenome graphs (that you can build with a tool such as panaroo)
*Thread Reply:* Oh! I didn't think about that! Thanks a lot 🙏 yes it's a mixture of closely related and more divergent so maybe I'll try the graph! ✨
🎉 New from the SPAAM Blog team (@Ele and me)! We really mean no stupid questions in this channel but we get it, sometimes you’re just too nervous to ask! Enter the “I’m too afraid to ask” form where you can post your anonymous questions! Ele and I will check in periodically and share submitted questions here… and maybe make a blog post out of the answers. You can also use the form to anonymously submit blog post ideas. It is pinned at the top of the channel whenever you need it! 🥳 Have fun!

Hi everyone! Has anyone ever made a Kaiju database including extra NCBI ID's for specific taxa (additionally to the "nr" db which one can specify)? Thanks!! 🙏
*Thread Reply:* I may know someone who has... but I'm waitnig for their answer. I seem to remember custom Kaiju DBs were complicated
*Thread Reply:* Omg thanks! Waiting excitingly! I've been pulling my hair over this
*Thread Reply:* I don't promise anything though 😬
*Thread Reply:* Nono no worries. A shred of hope that someone has an idea about this is better than nothing though
*Thread Reply:* You're in luck... sort of 😆
*Thread Reply:* https://hackmd.io/@jfy133/HJ3Pa4Mdo#Kaiju
*Thread Reply:* I guess you would need to add a few bits of R code to insert your extra specifi ctaxa

Hello! Does anyone have advice on long term storage of permafrost once it's been thawed? I have cores at -20C but I'm torn on whether to keep my subsamples at 4C or -20C. Ideally I would like to return to these subsamples in the future instead of resampling the core. Thank you!

*Thread Reply:* Best to use -20C for long-term storage of permafrost subsamples — they are a great substrate for microbial and fungal growth, which is only slowed at 4C.

There’s something I’ve been wondering about lately that I can’t seem to find a answer for, is there a way to change a value given to a directive in a process in a nf-core eager config with out having to write a whole config?
Say for example if I was using a default docker/singularity profile and I only wanted to change how long adapter removal can run for before it times out or how much memory it can use, and I wanted everything else to remain as default is there a flag I could pass in the command line to change this value?
*Thread Reply:* I am almost positive this all needs to be in a config file, rather than anything that could be passed as a parameter to the actual call of the pipeline. Sorry about that!
the benefit of this is that once you have your config file it will be usable across different runs and help with making sure that when you run it on your cluster you know it will work as you expect!
*Thread Reply:* here is the CLI info from nextflow itself, from what I can tell there is nothing that can be changed in terms of runtimes/resources for a job, etc. except with a config file https://www.nextflow.io/docs/latest/cli.html
*Thread Reply:* I had a feeling this would be the case based on what I read, oh well it was much more of a curiosity rather than an actual issue. Though I do wonder if you set the values as variables rather then hard coded numbers could you then override the default through passing a flag 🤔
I could definitely see a use case for such a feature
*Thread Reply:* Based on my understanding your OP, Ian is correct.
You can't change the resource directives via the cli.
You're not meant to need to do this. You should set these appropriately and/or dynamically once, and shouldn't have to touch it again (so least based on the nextflow concept). You don't want to pollute your reproducabile CLI command with extra infrastructure specific flags etc.
*Thread Reply:* So it would suggest if you're hitting cases where you run out of time (for example) your default is suboptimally set or you've not configured the auto retry correctly
*Thread Reply:* (if this makes sense)
Happy Monday! I present our first anonymous question for the community! Let’s help our anonymous friend out 💞
I know everyone is doing shotgun these days. But is there any update in processing 16S metagenomic data? I am currently using qiime2. Everyone in aDNA I asked so far dismissed amplicon sequencing altogether. So if anyone has an idea or works with that, I’d be infinitely grateful for helpful comments.
*Thread Reply:* Oofff this is a hard one... My question for the person would be do they know why we do shotgun?
*Thread Reply:* (maybe if they are watching they can reply on the anon form)
*Thread Reply:* Because the Reason why is amplicon primers are too long for most ancient reads, so the vast majority of reads you amplify will be modern
*Thread Reply:* (if they didn't know that already)
*Thread Reply:* It can be tough if you’re a student and the PI made the experimental design. But I think at that stage it would be important to discuss it with them and try to see if there could be any way to complement this analysis with some shotgun sequencing
*Thread Reply:* Yes exactly... (thus my initial question..)
*Thread Reply:* But I would argue there is 'no update' because it shouldn't work (or will be extremely difficult)
*Thread Reply:* Anonymous, I’m keeping an eye on the form so if you want to respond feel free to use the form again!
*Thread Reply:* (sorry to be so negative! But Like @Zoé Pochon says could definitely have a discussion to help come up with a plan to help get around some of the problems it poses [and why people are avoiding it]🙂 )
*Thread Reply:* https://www.nature.com/articles/srep16498
*Thread Reply:* Thanks @Meriam Guellil! Was looking for that until 👶 said no
*Thread Reply:* Hold up — is anon doing microbial 16S metabarcoding (with long insert lengths and hence problematic) or vertebrate 16S metabarcoding (with short insert lengths and suitable in some contexts)?
*Thread Reply:* Good poinT!
Thanks to everyone who answered our last anon. question. Here we have another for you metagenomics pros:
“I want to use SourceTracker but I don't know how to construct the database. Any of you know how? I want to include different sources, not only human-related microorganisms.”
Questions about SourceTracker have come up before so we would be really keen to run a blog post all about getting the software installed, creating databases and different parameters - let us know if you’d be interested in volunteering your expertise! Even if you don’t consider yourself an “expert”, we’d love to hear anecdotes about what’s worked for you and what hasn’t.
*Thread Reply:* There isn't a specific 'database' for Sourcetracker!
You just pick your 'sources' from whatever is appropriate for your question, and run both those and the sinks through a taxonomic classifier against the same database. The input is literally just an OTU table, and a metadata table saying which sample is a source, and which is a sink 🙂

*Thread Reply:* In case you want to use the MGnify biomes to retrieve sources from ENA this can be helpful https://github.com/genomewalker/get-biomes
*Thread Reply:* This is what I use to gather sources for the sourcetracking analyses
*Thread Reply:* You should start publishing or Zenodo'ing these 😉
*Thread Reply:* (note Zenodo has auto github archiving 😉 )
*Thread Reply:* Is coming a methods preprint with all these tools that make life easier
*Thread Reply:* I am compiling some of them here: https://github.com/aMG-tk
*Thread Reply:* Also if you use sourcetracker, I recommend using our extension with the diagnostics so you can evaluate how the MCs converge (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7100590/)

*Thread Reply:* I agree with everything James and Antonio have put here. I would just add that another consideration is to try to include sources that were prepared in a similar fashion as your data was created (e.g., shotgun, paired end vs single end, etc). It may also be useful to do some quality control on your sources before running Sourcetracker. Removing contaminant taxa in the sources. Another consideration may be to do a random forest or PCoA with only the source samples to check their homogeneity. I know that ST2 has an option that checks this but I think it would still be worth doing one of the two options above. Depending on the similarity of your environment sources, you may have several taxa that are present in multiple sources which can impact your downstream results.
*Thread Reply:* Exactly, get-biomes was designed with this in mind, to narrow down the potential sources that might fulfill your sink conditions. And as @Sterling Wright says, it is very important to explore and pre-process your abundance tables before ST. We also only use only damaged taxa for the sinks (when possible).
*Thread Reply:* We also used get-biomes to train @Camila Duitama decOM beyond oral microbiomes
*Thread Reply:* Was this the Kap København Formation paper@Antonio Fernandez-Guerra ?
*Thread Reply:* Yep, and other projects we are complementing ST with decOM
*Thread Reply:* and if you’re looking at human specific biomes, curatedMetagenomicsData is also a great resource (even with pre-computed metaphlan3 profiles)
Hi all!
I am interested in getting to know a bit more about human aDNA obtained in Colombia. I'd like to know a bit more about the outcomes of those publications, researchers (foreign and local) working on those samples, ethical issues, etc. Any suggestions?
Thanks!
*Thread Reply:* HAAM community for the rescue! @Thiseas C. Lamnidis (but he's sitting next to me atm so will reply later)
*Thread Reply:* https://haam-community.github.io/

*Thread Reply:* for local researchers, check the work of Andrea Casas-Vargas (mtDNA control region or hg only). Foreign, check this: https://www.sciencedirect.com/science/article/pii/S1040618220304961

*Thread Reply:* i’m not aware of paleogenomic studies or ethical discussions, but maybe Andrea Casas-Vargas can help?
Hiya, does anyone have any experience using mixer mills jars to generate powder in the lab? They are expensive so I want to make sure I am ordering something that will last!
Interested in hearing about what size jars people use? What material are they made of? How do you effectively clean the jars between samples?
Thanks! 🦴
*Thread Reply:* I’ve used one years ago for ancient DNA samples (Master thesis 2013, PCR-based stuff), my colleagues here use one to prepare bone samples for stable isotope analyses. For the DNA samples back then we used Zirconium oxide jars and a Retsch MM2 mill. We cleaned them with bleach, water and UV irradiated them. In my memory they were quite easy to clean.
*Thread Reply:* We use them and clean with bleach, dna off, ethanol & then UV them. They're pretty easy to clean but we do give them a good scrub in the bleach before the other steps. 🙂 Not sure which ones we actually use, but I think they're about 10cm long-ish??? and maybe 5cm in diameter, but this is from memory (I haven't drilled anything in nearly 2 years at this stage).
*Thread Reply:* Hi, I haven't used them in years but one thing I remember is that the polycarbonate cylinders were turning yellow and brittle-ish, possibly due to bleach and UV exposure.
*Thread Reply:* Thank you all for the helpful replies 🙂
Hi! I've got a memory of someone mentioning a while back that a fish genome had lots of adapters. Was it carp? I'm seeing it in extraction controls for my samples of sloth coprolites and from a layer of dung in the site (15 reads), then also in my samples (up to 130 reads or 0.02% of ids in a sample of soil, and up to 500 reads or 0.13% of ids in a coprolite) - If this is right, do you have a reference that discusses this?
*Thread Reply:* Yes, the carp genome (Cyprinus carpio) is contaminated with adapters.
*Thread Reply:* It was identified even earlier!!!
*Thread Reply:* https://twitter.com/baym/status/1712550914077372880


*Thread Reply:* I have used this as a teaching example on the importance of understanding the background of online genomic databases for many years now
Yes, Cyprinos carpio! https://dgg32.medium.com/carp-in-the-soil-1168818d2191

Hey lovely people! Does anyone have experience with using Kaiju for ancient data? If yes, how do you authenticate the hits? (Here I'm thinking something corresponding to the ED, ANI, breadth of coverage, ancient damage etc.??) Thanks!
*Thread Reply:* My understanding was it's not recommended as you translate to amino acid sequences, so you loose as unspecific all the Very short actually ancient reads during translation.
And then because you're dealing with amino acids can't do any of our normal validation
*Thread Reply:* Yes, I second this. The translation step would loose almost all reads shorter than 60 bp because there is limited resolution in assigning amino acid sequences with fewer than 20 aa.
*Thread Reply:* @James Fellows Yates you can use my emoji here… now we are getting good results down to 15 aa, and we will be pushing it down to 10aa in the near future with new sub matrices. We combine read-extension, with a fine tuned version of mmseqs2 and the E-M filtering from https://github.com/genomewalker/x-filter to maximise the information we can use from translated-searches (function and taxonomy)
*Thread Reply:* But that's not Kaiju is it ;)
Also, publish something then period can use it 🤣
*Thread Reply:* But exciting!
*Thread Reply:* It has been used for the 2-million year old stuff, at least is in biorxiv
*Thread Reply:* > But that’s not Kaiju is it 😉 No, I don’t think Kaiju can use other matrix than BLOSUM, which is not recommended for our type of data
*Thread Reply:* and also damage introduces artificial non-syn subs and stop codons which are not very suitable for methods like kaiju
*Thread Reply:* @James Fellows Yates @Alex Hübner @Antonio Fernandez-Guerra @Meriam Guellil @Carly Scott Follow up on the question from last time: does any of you happen to know any publications that mention this issue - especially maybe on the loss of specificity due to homologous peptide regions? But anything really. 🙇
*Thread Reply:* Internet is bad, so can't Google, but maybe @Raphael Eisenhofer's MALT blastx Vs maltn mode paper might discuss a littke? I think it was in peerj
*Thread Reply:* Not aDNA related but this might be useful https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03802-0 This is the approach we reimplemented in xfilter
Does anyone actually understand how metaDMG creates the output csv files? I have been looking at one example for a while now and can't figure it out. When I check the underlying lca files, every line is a unique read assigned to the lowest taxonomic level but in the metaDMG output file, several rows describe the same reads on different taxonomic levels (see attached). I don't really understand the rationale behind this and it makes the files a bit hard to work with but maybe I am missing something?

*Thread Reply:* From my understanding, this is because metaDMG calculates damage at all taxonomic levels. When moving one rank up, it gathers all daughter taxa and recalculate damage. In your example, from Gnathostomata and up there is no new taxon joining, therefore N_reads stays at 85.
*Thread Reply:* Ahh yes, you are right! I got too distracted by the fact that there is no obvious order of the rows and I was also expecting more reads on higher taxonomic levels but yes, when I sum up the reads of e.g. all classes, I get the clade reads etc. Thank you so much 🙂
Hey everyone 👋
We have another anonymous question, help a metagenomics pal out:
Hello everyone! We are working with lake sediments and we are wondering what to consider when preparing samples for dating. What do you usually do when macrofossils do not preserve well in your sediments? Have you ever used e.g. pollen extracts for AMS dating?
👀
*Thread Reply:* Sediments 🤔 @Stephanie Dolenz perhaps?
*Thread Reply:* I haven’t tried to date sediment samples yet, but perhaps @Kevin Nota?
*Thread Reply:* Mmm, I also never dated samples myself. Maybe @Pete Heintzman can help with this?
*Thread Reply:* Have not used pollen extracts for dating but it is possible. Suggest contacting the radiocarbon lab that you use about this.
Do you have any macrofossils at all? Even if they are very small/fragmentary, then these are usually datable (assuming you are looking at a Holocene record -- might not work well if going into MIS2 or MIS3 sediments, closer to the radiocarbon limit).
Depending on the sediment type, you could also try bulk sediment dating (especially of any organic layers). However, this assumes that there is very little to no carbonate present in the sediment (which is radiocarbon-dead and will give too old dates).
OP: feel free to DM me if you want to follow up! Cheers, Pete
*Thread Reply:* Was definitely hoping you would just tag someone else, @Pete Heintzman 🙂
*Thread Reply:* As a follow-up, I am sure that @Benjamin Vernot can offer some insight. 😉
*Thread Reply:* haha, not ME!
Hi everybody! I'm in the middle of extractions with Roche high-volume silica columns and realized I don't have buffer PE, which is what I usually use to wash the membrane. I do have the wash buffer that comes in the Roche kit and Qiagen buffer C5. I'm pretty sure they're all basically the same thing, but thought I'd ask others' opinions before doing anything! Thanks!
*Thread Reply:* Already talked to Ciara about this but a general FYI to any wet lab folks:
Ready-to-use wash buffer compositions: • Qiagen PE buffer: 10 mM Tris (pH 7.5) and 80% ethanol (unconfirmed but suspected composition) • Roche HPVNALV kit buffer: 2 mM Tris (pH 7.5), 20 mM NaCl, and 80% ethanol (confirmed composition) If anyone home-brews, make sure to use the correct Tris-HCl pH, as silica binding/release is highly sensitive to pH:

*Thread Reply:* Thank you again Pete! We ended up getting a pretty good DNA yield out of these. I might do a comparison of the different commercial and home brew wash buffers at some point just to see how different they are. Happy to eventually share the results with anyone who uses the Dabney extraction protocol and is also curious.
HI all! A researcher I worked with has a question and I was wondering whether anyone has any insights: "I am trying to calculate dN/dS ratios within-species for genes in S. sp. DD04 and S. sanguinis, and I have a few questions and was wondering if you could offer any insight. I have tables with the breadth and depth of coverage for each protein coding sequence in the annotated genome but am not sure how to go about sample/gene selection. What would be considered "sufficient coverage" of a gene to use in this analysis? Right now I have that minimum coverage of 3 is needed to call a variant, so I was thinking a depth of at least that, as well as breadth >95% or 100%.
I've seen that most programs (like CODEML and FUBAR) are guided by a phylogeny built with the genes of interest, which allows for finely characterizing selection pressures, including across different branches. However, given it is ancient DNA, I think this would be limited to genes that have sufficient coverage across all included samples. I also saw the program GenomegaMap which is phylogeny free and made for within-species estimation. Do you have any suggestions about which could be the more appropriate approach?"
*Thread Reply:* Hey @Abby Gancz , From my (short) experimentations, I found HyPhy nice to use https://hyphy.org/ (also online with https://datamonkey.org/
Regarding your gene inclusion thresholds, you can also look at the variant calling quality with bcftools and filter on that instead. (for example Q>=20 and allele support >= 3)
Hey all! I'm trying to use blastn on the command line and then visualise the results in megan, and I'm a bit stuck. I tried various things, but both the blast2rma tool and the megan app keep saying something like "Warning: Might not be a BLAST file in XML/SAM/BlastText format: 8511-37.blastn.out", while I do specify a specific output. What am I missing? Code I use to run blastn (for XML): blastn -query ${directory}/${sample}.collapsed.fasta -db nt -out ${sample}.blastn.out -outfmt 5 -numthreads $SLURMCPUSPERTASK
*Thread Reply:* Did you ever solve this @Meriam van Os?

Hi all, I was wondering if anyone here could help me with my specific issue regarding adding custom genomes into kraken2 🙂 I have been trying to incorporate two additional genomes of a species that is part of the standard library in kraken2 to the database I would use to analyse my asedDNA. What I’m trying to achieve is that these two additional reference genomes would show as subspecies in the final taxonomy. I attempted to do this by downloading the names.dmp and nodes.dmp files and adding these genomes to those files with custom taxIDs. However, it seems that the custom genomes are still not present at all in the final database. Would anyone have any idea what went wrong with this approach? Thanks!
*Thread Reply:* Could you provide all the commands you've used?
*Thread Reply:* Hi James,
Sure, thanks!
So at first I downloaded the taxonomy as follows:
kraken2-build --download-taxonomy --db JansedDNADB
And then the standard libraries, bacteria here as example:
kraken2-build --download-library bacteria --db JansedDNADB --threads 4
I then downloaded the names.dmp and nodes.dmp files and added the following lines there as follows:
9999999 | 69293 | subspecies | GA | 10 | 1 | 1 | 1 | 2 | 1 | 1 | 0 | | 9999998 | 69293 | subspecies | GA | 10 | 1 | 1 | 1 | 2 | 1 | 1 | 0 | |
9999999 | Gasterosteus aculeatus freshwater | | scientific name | 9999998 | Gasterosteus aculeatus marine | | scientific name |
After uploading these files back to the taxonomy folder of the database I added the custom taxIDs on the fasta files of the custom genomes
>Freshwater|kraken:taxid|9999999 >Marine_fjord|kraken:taxid|9999998
These genomes also had chromosomes indicated with >chr** so I erased all the lines starting with > except the first one.
sed -i ‘2,${/^>/d}’ gasAcu1.fa sed -i ‘2,${/^>/d}’ fjord.fa
And then I added the genomes:
kraken2-build --add-to-library gasAcu1.fa --db JansedDNADB kraken2-build --add-to-library fjord.fa --db JansedDNADB
And finally I just ran the build command:
kraken2-build --build --db JansedDNADB --threads 40
The final database works fine except it just seems that my custom genomes are not there at all. Did I skip some step or do something wrong?
*Thread Reply:* How do you validate that the custom genomes are not in there?
*Thread Reply:* I noticed the same issue for my custom built db @Jan Laine
*Thread Reply:* No solution? Because yeah I agree the commands look ok to me :(
*Thread Reply:* I’m not sure neither, but that seems to be an issue that has been reported on their github https://github.com/DerrickWood/kraken2/issues?q=is%3Aissue+is%3Aopen+custom
*Thread Reply:* From my understanding, it works to add genomes to a completely new database, but if you want to use these pre-selected databases, it doesn’t work. But it’s not clearly documented anywhere
*Thread Reply:* Ahhh that would make sense, I had no issues with this on a custom database for nf-core/taxprofiler, but that was pure xustom
*Thread Reply:* Okey thanks a lot for looking into this, I really appreciate it! To validate that they are not in there I just first assumed so since they got no hits even though they should with the samples i tested it. I also downloaded the report and tried to search the custom taxIDs without any hits. But good to know that this seems to be the issue then that it needs to be completely custom, maybe I can work with that info! 🙂
*Thread Reply:* You can check with the inspect command
*Thread Reply:* But yeah, that’s not an ideal situation
*Thread Reply:* Ah ok. I don't think just not finding hits is necessaily the best way, could be they get moved up the LCA because the genomes are similar? Unless you're looking at the per read hits...
Agree with maxime better to investigate the database itself
*Thread Reply:* Okay thanks, I’ll check that as well!
*Thread Reply:* Seconding that, in my experience I had to start from scratch, downloading all the genomes and running build, when I wanted to just add a couple taxa of interest to the standard database and it was a pain 😕 I wonder if you could avoid the computational hassle by creating a smaller custom database of just that genus/species including your subspecies? And then run just the reads ID’d as the species of interest from the standard library against the custom database? I need to think through that a bit more though
*Thread Reply:* Do other users of Kraken2 have notice the same behavior ? (@Nikolay Oskolkov maybe ?)
*Thread Reply:* Seems like the behaviour also reported here: https://github.com/DerrickWood/kraken2/issues/538
<a href="https://github.com/DerrickWood/kraken2">DerrickWood/kraken2</a>

Hi all, I have been thinking about damage profiles and noticed that while the frequency of C->T substitutions due to deamination is significantly higher at the ends of DNA fragments, one can (often) observe a persistent (very low) frequency of these substitutions observed internally within the fragments, e.g. converging at around 1%. Now I wonder why that is, because non-typical ancient DNA substitutions often have a baseline of 0%. Could it be that C->T substitutions are indeed occurring at a low level throughout the DNA fragments, or might this be influenced by the inherent mutation rates of cytosine and thymine? Or could this pattern be an artifact of the damage estimation methods we use to estimate DNA damage? Any thoughts? Thanks! :)

*Thread Reply:* Are these bacterial fragments or Eukaryotic?
*Thread Reply:* Could also be that you’re mapping to a further away reference
*Thread Reply:* Is it only C->T, or also other substitutions ?
*Thread Reply:* Check the 5' G-->A. If that is at the same level of the mid-molecule C-->T, then suggestive of evolutionary divergence from reference.
Or were these sequenced from single-stranded DNA libraries?
*Thread Reply:* And you don’t see it as much on human aDNA damage, because well, usually you know what species/“strain” you’re looking at 😉
*Thread Reply:* It was more of an overall observation, so I was not talking about my own data. I found examples of both, human and bacterial samples. So I guess you would agree, that it is a common thing one can observe in samples? 🙂
*Thread Reply:* Good question, thank you for asking that 😉 I've come across this also some times and wondered
*Thread Reply:* Wouldn't these be the traces of deaminated CpG positions? They would show up as C-->T as well, wouldn't they?
*Thread Reply:* That’s what I was thinking of it was eukaryotic DNA. There could be a variety of reasons depending on the context, library preparation, and sequencing.

Hi everyone, I am looking for help regarding the use of KrakenUniq. Does anyone have experience with this software? I am planning to run samples containing between 19.000.000 and 40.000.000 reads (150PE) using the MicrobialDB (384GB). According to a first trial and my estimations, each sample will take between 10 to 20 days to be classify!!! I am copying the DB to the local disk before the run and use the script below. The same samples using Kraken2 only need a few hours to be classify. Does anybody have any suggestion? I thank you all in advance. Nathalie
SBATCH --ntasks=1
SBATCH --cpus-per-task=1
SBATCH --mem-per-cpu=1G
SBATCH --partition=batch
echo Start job and use local scratch date ml releases/2022a ml KrakenUniq/1.0.3-GCC-11.3.0
datadir=$LOCALSCRATCH/KrakenUniq uploaddir=./Upload echo Copy data base in local disk $datadir mkdir -p $datadir srun cp ./KrakenUniq/kuniqmicrobialdb.kdb.20200816.tgz ./KrakenUniq/database.kdb $datadir/. cd $datadir echo Extract data srun tar -xzf kuniqmicrobialdb.kdb.20200816.tgz cd -
files=( "sample1.pair1.truncated.fq.gz" # input file 1 "sample1.pair2.truncated.fq.gz" # input file 2 "sample1krakenuniqClass" # out file "sample2.pair1.truncated.fq.gz" "sample2.pair2.truncated.fq.gz" "sample2krakenuniqClass" )
ArrayLength=${#files[@]} n=1 for ((i = 0 ; i < ArrayLength ; i++ )); do jobname="${SLURMJOBNAME}-$n" filein1=${files[$i]} filein2=${files[(++i)]} fileout=${files[(++i)]} date echo Start task $n ...
srun -J "${jobname}" krakenuniq --db $datadir --paired --classified-out classifiedsequences-${jobname}.txt ${uploaddir}/${filein1} ${uploaddir}/${filein2} > ${uploaddir}/${fileout}-${job_name}
echo "End tasks $n" date ((n++)) done
echo End all job echo Remove local data srun rm -rf $data_dir date
*Thread Reply:* Hmm, KrakenUniq definitely takes longer than Kraken2 but not that much! Are you able to increase the memory for the job?
*Thread Reply:* That is what I was going to recommend too. Not sure what capacity of RAM that you have but maybe you can change --mem to a larger number than the default in your current system?
*Thread Reply:* Exactly, give it one of the largest nodes and it will run much faster. I’m using 1TB nodes and it runs normally in less than a day
*Thread Reply:* @Nathalie Suarez Gonzalez given identical RAM available (for example, a 512GB compute node for your 384GB microbial DB), Kraken2 will be (much) faster than KrakenUniq. However, if you have only e.g. 256GB node available, Kraken2 would not be possible to run with your 384GB DB, but KrakenUniq can be run, it will be slow though, i.e. slower than if you had 512GB of RAM available
*Thread Reply:* Also @Nathalie Suarez Gonzalez, KrakenUniq starts very slow, but accelerates dramatically towards the end (for unclear reasons, I guess is starts with longest reads first, and quickly maps the shortest reads at the end). From my experience, provided that you have enough RAM to accommodate your 384GB microbial DB (for example, if you have a 512GB compute node), and you enable multi-threading in KrakenUniq, 19 mln - 40 mln reads should not take more than 6-8 hours.
*Thread Reply:* Thank you very much for your input 🙂

Hey everyone, I am new to the topic of metagenomics and have many questions regarding this topic. I would like to filter kraken report based on a number of unique k-mers before deciding what bacteria or virus to focus on. I do not want to miss anything important but also would like to remove potential noise from my kraken report. What would be appropriate lower threshold for number of k-mers to extract? Some viruses have very small genomes so I cannot expect lots of unique k-mers for those...
*Thread Reply:* We use nuniquekmers=1000 in aMeta, this works well for bactetial genomes, but it is probably too conservative for viruses. I believe, nuniquekmers / Lgenome is a better filter, but I do not have a good threshold in mind. I would plot distributions of nuniquekmers / Lgenome for each sample (which will hopefully be bimodal) before deciding on exact threshold
*Thread Reply:* Yes, thank you, I read your article on aMeta and that default threshold sounds to hight, so that I why I decided to ask. Ok, I like very much your suggestion, but is there genome ref length stated somewhere in the kraken report? I have kraken2 report so should be similar to KrakenUnique.
*Thread Reply:* Otherwise, it would be a pain to look up every genome in the data base to fetch its length.
*Thread Reply:* Genome lengths you have to compute yourself by something like ”wc -c ref.fa”
*Thread Reply:* Thank you Nikolay, I will give it a try:)
*Thread Reply:* I like to look at the ratio between k-mers and reads for shotgun data.
*Thread Reply:* It’s a good indicator that the reads are distributed randomly over the genome if there are much more k-mers than reads. If there are more reads than k-mers it means the reads are probably stacking (or that you sequenced a loooot or captured)
*Thread Reply:* I’ve came up with this for KrakenUniq https://maximeborry.com/post/kraken-uniq/
*Thread Reply:* For kraken2 it’s slightly different, because it doesn’t compute a “coverage”
*Thread Reply:* I am using results from Kraken2, but is has 8 columns, so assuming the same ouput as KrakenUniq...
*Thread Reply:* For Kraken2, I worked out something similar, a score S where a high values S is more likely to be false positive, and low values of S is less likely to be false positive.
With d = nb_minimizer/nb_unique_minimizer and r=nb_reads

*Thread Reply:* Btw @Maxime Borry, regarding the blog post about the new e-score, do you have a suggestion of threshold for it as well?

*Thread Reply:* (edit: x axis more readable)
*Thread Reply:* @Zoé Pochon it’s the tricky balance between precision and recall, but looking towards the end of my blog post, you can see a range of values that would work
Hi everyone, fastq files demultiplexed are ready to use the aMeta pipelines, right?
*Thread Reply:* and can it run in a standard laptop? (or better in university's hpc?)
*Thread Reply:* Hey Yuti! Yes I think so! However, if it is paired-end data, you still have to merge it first cause we don't have that option. It's made to be ran on an hpc server. If you search the github page, you'll see a link to a suggested way to set it up on hpc servers.
*Thread Reply:* Hi @Yuti Gao, yes, raw reads (demultiplexed) in fastq-format are the standard input for aMeta. You can install and test aMeta on your laptop, however, for real world applications you will have to use big databases (we published a bunch together with aMeta paper, and you can find the links at the very bottom of README here https://github.com/NBISweden/aMeta). The big databases can efficiently be handled on an HPC. And the size (and RAM requirements) of the databases are not a peculiarity of aMeta itself but the metagenomic analysis in general which I doubt can be run on a laptop (any metagenomic analysis) 🙂
*Thread Reply:* thanks @Zoé Pochon, yes, it is pair-end data, do you know any written pipelines to do the merge?
*Thread Reply:* thanks @Nikolay Oskolkov, I was following the textbook section about aMeta, https://www.spaam-community.org/intro-to-ancient-metagenomics-book/ancient-metagenomic-pipelines.html#what-is-ameta, they looks like the same, is it?
*Thread Reply:* @Yuti Gao yes, the textbook chapter is good to follow. Regarding merging overlapping PE reads I could tecommend fastp or SeqPrep
*Thread Reply:* Otherwise simply ”cat R1.fastq.gz R1.fastq.gz > merged.fastq.gz” would also work as input for aMeta
*Thread Reply:* cat R1.fastq.gz R2.fastq.gz > merged.fastq.gz?
*Thread Reply:* I would go with fastp so that you merge potentially overlapping pairs of reads together instead of just concatenating the files in one file.
*Thread Reply:* when I run to test the installation,
cd .test
./runtest.sh -j 4
get an error,
Error in rule KrakenUniq:
krakenuniq: database ("resources/KrakenUniq_DB") does not contain necessary file database.kdb
Shutting down, this might take some time. Exiting because a job execution failed. Look above for error message Complete log: .snakemake/log/2023-11-21T121632.600005.snakemake.log
do you have any suggestions?
thank you!
*Thread Reply:* @Yuti Gao could you please post the log-file .snakemake/log/2023-11-21T121632.600005.snakemake.log here?
*Thread Reply:* @Yuti Gao could you please do
- rm -rf .test/results
- rm .test/resources/KrakenUniq_DB/database**
and restart the testrun via
./runtest.sh -j 4?
It looks like you tried to execute it a few times after something has failed in the first run
*Thread Reply:* (aMeta) [yuga3894@c3cpu-c11-u3-3 .test]$ rm .test/resources/KrakenUniqDB/database rm: cannot remove '.test/resources/KrakenUniqDB/database': No such file or directory (aMeta) [yuga3894@c3cpu-c11-u3-3 .test]$ cd resources/ (aMeta) [yuga3894@c3cpu-c11-u3-3 resources]$ ls accession2taxid.map ref.fa.2.bt2l ref.fa.rev.2.bt2l KrakenUniqDB ref.fa.3.bt2l samples.tsv pathogenomesFound.tab ref.fa.4.bt2l seqid2taxid.pathogen.map ref.fa ref.faBOWTIE2BUILD.log ref.fa.1.bt2l ref.fa.rev.1.bt2l (aMeta) [yuga3894@c3cpu-c11-u3-3 resources]$ cd KrakenUniqDB (aMeta) [yuga3894@c3cpu-c11-u3-3 KrakenUniq_DB]$ ls library seqid2taxid.map taxonomy
*Thread Reply:* looks like I don't have database within KrankenUniq
*Thread Reply:* @Yuti Gao please note the asterisk ** here: rm .test/resources/KrakenUniq_DB/database**
*Thread Reply:* @Yuti Gao good, please rerun the testrun
*Thread Reply:* I'm not in front of my computer, but I would say go into the .test folder, open the script (file finishing with .sh) and run what had to be run within the condition by hand. Sometimes the condition for the first run doesn't seem to activate depending on the environment to my experience.
*Thread Reply:* tried both, get same error when I do ./runtest.sh -j 4, no red error when I just do bash runtest.sh, but a lot of "No validator" and missing metadata, what should be the output?
*Thread Reply:* Running workflow... snakemake --use-conda --conda-frontend mamba --show-failed-logs --conda-cleanup-pkgs cache -s ../workflow/Snakefile Error: you need to specify the maximum number of CPU cores to be used at the same time. If you want to use N cores, say --cores N or -cN. For all cores on your system (be sure that this is appropriate) use --cores all. For no parallelization use --cores 1 or -c1. <io.TextIOWrapper name='<stderr>' mode='w' encoding='utf-8'> Generating report... snakemake -s ../workflow/Snakefile --report --report-stylesheet ../workflow/report/custom.css No validator found for JSON Schema version identifier 'http://json-schema.org/draft/2020-12/schema#' Defaulting to validator for JSON Schema version 'https://json-schema.org/draft/2020-12/schema' Note that schema file may not be validated correctly. Excluding samples 'foobar' from analysis Restricting analysis to samples 'foo','bar' No validator found for JSON Schema version identifier 'http://json-schema.org/draft/2020-12/schema#' Defaulting to validator for JSON Schema version 'https://json-schema.org/draft/2020-12/schema' Note that schema file may not be validated correctly. Changing directory from /projects/yuga3894/aMeta/.test to /projects/yuga3894/aMeta/workflow Changing directory back to /projects/yuga3894/aMeta/.test Building DAG of jobs... Creating report... Missing metadata for file results/MULTIQC/multiqcreport.html. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/BOWTIE2/foo/AlignedToBowtie2DB.bam. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/BOWTIE2/foo/AlignedToBowtie2DB.bam.bai. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/BOWTIE2/bar/AlignedToBowtie2DB.bam. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/BOWTIE2/bar/AlignedToBowtie2DB.bam.bai. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MAPDAMAGE/foo. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/krakenuniq.output.filtered. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/krakenuniq.output.pathogens. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/taxID.pathogens. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/taxID.species. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/krakenuniq.output. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/sequences.krakenuniq. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MAPDAMAGE/bar. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/krakenuniq.output.filtered. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/krakenuniq.output.pathogens. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/taxID.pathogens. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/taxID.species. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/krakenuniq.output. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/sequences.krakenuniq. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/krakenuniq.output.filteredtaxIDskmers1000.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/sequences.krakenuniqkmers1000.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/sequences.krakenuniqkmers1000.krona. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/foo/taxonomy.krona.html. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/krakenuniq.output.filteredtaxIDskmers1000.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/sequences.krakenuniqkmers1000.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/sequences.krakenuniqkmers1000.krona. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQ/bar/taxonomy.krona.html. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTABUNDANCEMATRIXSAM. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTABUNDANCEMATRIXSAM/maltabundancematrixsam.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTQUANTIFYABUNDANCE/foo. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTQUANTIFYABUNDANCE/foo/samcounts.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALT/foo.trimmed.rma6. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALT/foo.trimmed.sam.gz. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTDB/seqid2taxid.project.map. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTDB/seqids.project. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTDB/project.headers. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTDB/library.project.fna. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTDB/maltDB.dat. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQABUNDANCEMATRIX. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQABUNDANCEMATRIX/uniquespeciestaxidlist.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQABUNDANCEMATRIX/uniquespeciesnameslist.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQABUNDANCEMATRIX/krakenuniqabundancematrix.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/KRAKENUNIQABUNDANCEMATRIX/krakenuniqabsoluteabundanceheatmap.pdf. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTQUANTIFYABUNDANCE/bar. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTQUANTIFYABUNDANCE/bar/samcounts.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALT/bar.trimmed.rma6. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALT/bar.trimmed.sam.gz. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTABUNDANCEMATRIXRMA6. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/MALTABUNDANCEMATRIXRMA6/maltabundancematrixrma6.txt. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/AUTHENTICATION/foo/.extracttaxidsdone. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/AUTHENTICATION/bar/.extracttaxidsdone. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/overviewheatmapscores.pdf. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/AUTHENTICATION/.foodone. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Missing metadata for file results/AUTHENTICATION/.bardone. Maybe metadata was deleted or it was created using an older version of Snakemake. This is a non critical warning. Downloading resources and rendering HTML. Report created: report.html.
*Thread Reply:* @Yuti Gao did you remove the .test/results prior to rerunning the testrun?
*Thread Reply:* also, after I installed aMeta in hpc i just see there is a section "Environment module configuration" in the github, should I do this part before I run the test
*Thread Reply:* @Yuti Gao no, wait a bit with the environment module configuration. Can you please show me the content of .test/results?
*Thread Reply:* (aMeta) [yuga3894@c3cpu-c11-u19-3 results]$ ls CUTADAPTADAPTERTRIMMING FASTQCAFTERTRIMMING FASTQCBEFORETRIMMING
*Thread Reply:* @Yuti Gao I am afraid it still did not work 😞 Could you please delete aMeta installation like this:
- rm -rf aMeta #remove the whole aMeta folder
- conda remove -n aMeta --all #remove aMeta environment and just redo everything from scratch, i.e. cloning of aMeta repo, creating aMeta environment and running the testrun. And please post here the very first issue you encounter (the log-file would be ideal)
*Thread Reply:* @Yuti Gao sorry that you encountered this problem. When running the test file for the first time, keep an eye open to see if it says something like :"This looks like the first run". If not it means it is somehow not gonna create the necessary test files before running the test.
*Thread Reply:* I reinstalled it, notice this message:
Krona installed. You still need to manually update the taxonomy databases before Krona can generate taxonomic reports. The update script is ktUpdateTaxonomy.sh. The default location for storing taxonomic databases is /projects/yuga3894/software/anaconda/envs/aMeta/opt/krona/taxonomy
should I run ktUpdateTaxonomy.sh before I run the test?
*Thread Reply:* I did get the first run message,
(aMeta) [yuga3894@c3cpu-c15-u9-1 .test]$ ./runtest.sh -j 4 This looks like the first test run... Installing bioconda packages...
... Your conda installation is not configured to use strict channel priorities. This is however crucial for having robust and correct environments (for details, see https://conda-forge.org/docs/user/tipsandtricks.html). Please consider to configure strict priorities by executing 'conda config --set channel_priority strict'.
and looks like eveything goes well until
MissingOutputException in rule KrakenUniq in file /projects/yuga3894/aMeta/workflow/rules/krakenuniq.smk, line 1: Job 16 completed successfully, but some output files are missing. Missing files after 5 seconds. This might be due to filesystem latency. If that is the case, consider to increase the wait time with --latency-wait: results/KRAKENUNIQ/bar/sequences.krakenuniq
*Thread Reply:* Hi @Yuti Gao, thank for the very detailed reporting, a few comments:
- The "no validator found" warning is fine, please ignore it
- The "Krona installed. You still need to manually update the taxonomy" message is also not very relevant for you at this stage (it will become important later), so please ignore it as well.
- The message "Your conda installation is not configured to use strict channel priorities" should also be ignored. Snakemake is ultra-verbose 🙂
- The log-file from the testrun you sent does not seem to be the log from the very first run. I would like to see the yellow messages regarding installing environments, something like:
Creating conda environment ../workflow/envs/malt.yaml... Downloading and installing remote packages. - Immediately after the yellow messages about installing environments, there will be some white text concerning building a KrakenUniq_DB on the fly, something like:
Building krakenuniq data
Kraken build set to minimize disk writes.
Finding all library files
Found 1 sequence files (**.{fna,fa,ffn,fasta,fsa}) in the library directory.
Creating k-mer set (step 1 of 6)...
This step seems to fail for you and I need to see the log-file to understand why.
In summary, could you please do again:
- rm -rf aMeta #remove the whole aMeta folder
- conda remove -n aMeta --all #remove aMeta environment
- git clone https://github.com/NBISweden/aMeta
- cd aMeta
- mamba env create -f workflow/envs/environment.yaml
- conda activate aMeta
- cd .test
- ./runtest.sh -j 4 and send me the log-file? Please do not restart the testrun if you suspect that something is not going right, I need to see the log-file from the very first run (failed or successful). The log-file can be found following the message at the very end, something like this "Complete log: .snakemake/log/2023-11-22T091420.873756.snakemake.log"
*Thread Reply:* Is there an .initdb file in your .test folder after running the test? It would mean that it really went through the first run condition all the way. If not, I would take the code from the condition and run it on the terminal, from within the .test folder :
*Thread Reply:* ```echo "This looks like the first test run... Installing bioconda packages..." snakemake --use-conda --show-failed-logs -j 2 --conda-cleanup-pkgs cache --conda-create-envs-only -s ../workflow/Snakefile
source $(dirname $(dirname $CONDA_EXE))/etc/profile.d/conda.sh
##############################
# Krakenuniq database
##############################
echo Building krakenuniq data
env=$(grep krakenuniq .snakemake/conda/**yaml | awk '{print $1}' | sed -e "s/.<yaml://g>")
conda activate $env
krakenuniq-build --db resources/KrakenUniq_DB --kmer-len 21 --minimizer-len 11 --jellyfish-bin $(pwd)/$env/bin/jellyfish
conda deactivate
##############################
# Krona taxonomy
##############################
echo Building krona taxonomy
env=$(grep krona .snakemake/conda/**yaml | awk '{print $1}' | sed -e "s/.<yaml://g>" | head -1)
conda activate $env
cd $env/opt/krona
./updateTaxonomy.sh taxonomy
cd -
conda deactivate
##############################
# Adjust malt max memory usage
##############################
echo Adjusting malt max memory usage
env=$(grep hops .snakemake/conda/**yaml | awk '{print $1}' | sed -e "s/.<yaml://g>" | head -1)
conda activate $env
version=$(conda list malt --json | grep version | sed -e "s/\"//g" | awk '{print $2}')
cd $env/opt/malt-$version
sed -i -e "s/-Xmx64G/-Xmx3G/" malt-build.vmoptions
sed -i -e "s/-Xmx64G/-Xmx3G/" malt-run.vmoptions
cd -
conda deactivate
touch .initdb```
*Thread Reply:* @Nikolay Oskolkov I found my terminal record after this:
Creating conda environment ../workflow/envs/malt.yaml...
Downloading and installing remote packages.
if this doesn'r work, I will rerun the installation
*Thread Reply:* @Zoé Pochon thanks, is the .initdb file directly under the /test folder, looks I don't have it then [yuga3894@login13 .test]$ ls benchmarks config data logs report.html resources results runtest.sh
*Thread Reply:* ls -a To see the hidden files that begin with a dot
*Thread Reply:* @Yuti Gao from the the log-file you posted it seems your KrakenUniq_DB was correctly built, so it seems to me that you should have had the database-files, which you previously reported missing. I suggest we have a clean run, so could you please do these steps and post the log-file here:
In summary, could you please do again:
- rm -rf aMeta #remove the whole aMeta folder
- conda remove -n aMeta --all #remove aMeta environment
- git clone https://github.com/NBISweden/aMeta
- cd aMeta
- mamba env create -f workflow/envs/environment.yaml
- conda activate aMeta
- cd .test
- ./runtest.sh -j 4
*Thread Reply:* (aMeta) [yuga3894@c3cpu-a5-u28-1 .test]$ ls benchmarks config data logs report.html resources results runtest.sh
*Thread Reply:* just remove everythign and reinstall, looks like I got the same error
*Thread Reply:* Thank you @Yuti Gao, this looks good, please do not tweak anything in aMeta right now, let me investigate it a bit. Your KrakenUniq_DB building process looks fine, so there must be something wrong with the KrakenUniq run itself. Could you please post here the log-file located in aMeta/.test/logs/KRAKENUNIQ/bar.log?
*Thread Reply:* Ok, thank you @Yuti Gao, this explains it!
NOTE: No need to use --gzip-compressed or --bzip2-compressed anymore, format is detected automatically.
NOTE: No need to use --fasta-input or --fastq-input anymore, format is detected automatically.
/projects/yuga3894/aMeta/.test/.snakemake/conda/74c9aeceba45102acd1e47590c34ff6d_/share/krakenuniq-1.0.4-1/libexec/classify -d resources/KrakenUniq_DB/database.kdb -i resources/KrakenUniq_DB/database.idx -t 4 -o results/KRAKENUNIQ/bar/sequences.krakenuniq -c -M -r results/KRAKENUNIQ/bar/krakenuniq.output -a resources/KrakenUniq_DB/taxDB -p 12
classify: thread count exceeds number of processors
*Thread Reply:* I believe you do not have 4 processors available, so you should run it as
./runtest.sh -j 1
*Thread Reply:* should I talk to hpc staff to get 4?
*Thread Reply:* Are you using login node of your HPC or did you reserve a node (with several CPUs) for running aMeta?
*Thread Reply:* right now you should just try ./runtest.sh -j 1. If this solves the issue, we can talk how to run aMeta on real data, then you certainly need nodes with many CPUs
*Thread Reply:* I think it's login node now, I did get an account to reserve a node, although I don't know how to do it now
*Thread Reply:* Error in rule BuildMaltDB: jobid: 22 input: results/KRAKENUNIQABUNDANCEMATRIX/uniquespeciestaxidlist.txt output: results/MALTDB/seqid2taxid.project.map, results/MALTDB/seqids.project, results/MALTDB/project.headers, results/MALTDB/library.project.fna, results/MALTDB/maltDB.dat log: logs/BUILDMALTDB/BUILDMALTDB.log (check log file(s) for error details) conda-env: /projects/yuga3894/aMeta/.test/.snakemake/conda/dcf57682cc9d8eaf05305773b2b3d994_ Logfile logs/BUILDMALTDB/BUILDMALTDB.log:
*Thread Reply:* Ok, it seems that unfortunately your login node provides only very little RAM, something like 4-8 GB, which is not enough even for building this very small Malt database. @Yuti Gao would it be possible to to book a node with more resources?
*Thread Reply:* alternatively, your laptop should have more resources compared to this HPC login node 🙂 so you might want to try installing aMeta on your laptop and learn the installation process
*Thread Reply:* ohhh, ok then I would talk to hpc after people come back from this thanksgiving holiday, yes, I can book a node with more RAM.
*Thread Reply:* thank you Nikolay for the help these days!
*Thread Reply:* You are welcome @Yuti Gao, and I am very sorry for all the troubles! Eventually, it looks like the problem with KrakenUniq was due to the lack of available threads on the login node (so we should put ./runtest.sh -j 1 to aMeta's README in order to avoid it in the future), and the problem with Malt was due to the lack of RAM on the login node.
Strategically, I would encourage you to learn how to book nodes with a lot of RAM in your HPC system, since aMeta will require much more RAM to use real databases on real-world data. Once you have booked a node with some reasonable RAM (at least 128-256GB, preferably more), please DM me and I will do my best to help you
Hello! More of a conda issue than an aMeta one so I am not going to bother Zoé and Nikolay directly in case anyone else has dealt with this error:
ModuleNotFoundError in file /scratch/shreya23/aMeta/workflow/rules/common.smk, line 6:
No module named 'pytz.tzinfo'
…but when I do conda list it seems like pytz is installed:
pytz 2023.3.post1 pyhd8ed1ab_0 conda-forge
and removing/uninstalling python and pytz doesn’t seem to fix it!
*Thread Reply:* @Shreya strange, I have never seen this error, and it is puzzling that the line 6 in common.smk does not seem to directly deal with pytz

*Thread Reply:* When did this error popup? During installation or testrun or later?
*Thread Reply:* During the test run! No issues popped up at installation.
*Thread Reply:* @Shreya are you sure you activate aMeta conda environment? If yes, what if you pip install pytz ?
*Thread Reply:* I’m pretty sure I activated it, but let me try again!
*Thread Reply:* Unfortunately pip install pytz gives me the same error. I’ve got 2 separate conda installations I’m trying, one I did myself and one our admin installed on the cluster, and I get the same error for both. Here’s the full error:

*Thread Reply:* Perhaps this is a question for cluster admin then?
*Thread Reply:* Yes, the line 6 in common.smk is importing pandas, and the pytz error has something to do with pandas from the screenshot you posted. Can you at all run import pandas if you start Python? Looks like something is wrong with pandas installation
*Thread Reply:* (aMeta) [shreya23@cri22cn094 .test]$ python
Python 3.10.13 | packaged by conda-forge | (main, Oct 26 2023, 18:07:37) [GCC 12.3.0] on linux
Type "help", "copyright", "credits" or "license" for more information.
>>> import pandas
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
File "/home/shreya23/.conda/envs/aMeta/lib/python3.10/site-packages/pandas/__init__.py", line 46, in <module>
from pandas.core.api import (
File "/home/shreya23/.conda/envs/aMeta/lib/python3.10/site-packages/pandas/core/api.py", line 1, in <module>
from pandas._libs import (
File "/home/shreya23/.conda/envs/aMeta/lib/python3.10/site-packages/pandas/_libs/__init__.py", line 18, in <module>
from pandas._libs.interval import Interval
File "interval.pyx", line 1, in init pandas._libs.interval
File "hashtable.pyx", line 1, in init pandas._libs.hashtable
File "missing.pyx", line 1, in init pandas._libs.missing
File "/home/shreya23/.conda/envs/aMeta/lib/python3.10/site-packages/pandas/_libs/tslibs/__init__.py", line 39, in <module>
from pandas._libs.tslibs.conversion import localize_pydatetime
File "conversion.pyx", line 1, in init pandas._libs.tslibs.conversion
File "offsets.pyx", line 1, in init pandas._libs.tslibs.offsets
File "timestamps.pyx", line 1, in init pandas._libs.tslibs.timestamps
File "timedeltas.pyx", line 1, in init pandas._libs.tslibs.timedeltas
File "timezones.pyx", line 32, in init pandas._libs.tslibs.timezones
ModuleNotFoundError: No module named 'pytz.tzinfo'
*Thread Reply:* And then if I deactivate aMeta and just try on base:
(base) [shreya23@cri22cn094 .test]$ python
Python 3.10.11 | packaged by conda-forge | (main, May 10 2023, 18:58:44) [GCC 11.3.0] on linux
Type "help", "copyright", "credits" or "license" for more information.
>>> import pandas
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
ModuleNotFoundError: No module named 'pandas'
*Thread Reply:* Oho… seems like something is up here! Thank you so much for helping me debug
*Thread Reply:* @Shreya I would first try to install pandas in your base-environment with pip install pandas and then just delete aMeta installation like this:
- rm -rf aMeta #remove the whole aMeta folder
- conda remove -n aMeta --all #remove aMeta environment and just redo everething from scratch, i.e. cloning of aMeta repo, creating aMeta environment and running the testrun. This is just to be on a safe side
*Thread Reply:* Okay!! Thank you so much Nikolay!! Will give this a shot and keep you posted!!
*Thread Reply:* Yes @Shreya, we can have a zoom meeting with screen sharing if needed, and I will try to do my best to help you
*Thread Reply:* Thank you!! I really appreciate it! And I know it is rather late in Sweden right now!
*Thread Reply:* Hi Nikolay!! Great news!! Installing pandas in base worked and now the workflow is starting!!
There is a new issue that I’m hoping is an easier fix? In running the test script I now get this error:
MissingOutputException in rule Bowtie2_Index in file /scratch/shreya23/aMeta/workflow/rules/align.smk, line 1:
Job 9 completed successfully, but some output files are missing. Missing files after 5 seconds. This might be due to filesystem latency. If that is the case, consider to increase the wait time with --latency-wait:
resources/ref.fa.1.bt2l
resources/ref.fa.2.bt2l
resources/ref.fa.3.bt2l
resources/ref.fa.4.bt2l
resources/ref.fa.rev.1.bt2l
resources/ref.fa.rev.2.bt2l
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
I did edit the run script to add --latency-wait 60 (is that not long enough?) and got the same error. The files in resources are:
accession2taxid.map
KrakenUniq_DB
pathogenomesFound.tab
ref.fa
ref.fa.1.bt2
ref.fa.2.bt2
ref.fa.3.bt2
ref.fa.4.bt2
ref.fa_BOWTIE2_BUILD.log
ref.fa.rev.1.bt2
ref.fa.rev.2.bt2
samples.tsv
seqid2taxid.pathogen.map
so I am wondering if I need to rename the .bt2 files. to .bt2l? Thank you!!
*Thread Reply:* Hi @Shreya, great that it worked with pandas and, yes, the Bowtie2_Index seems to be easier to fix. Could you please send me the file ref.fa_BOWTIE2_BUILD.log from ./test/resources? Please do not modify the --latency-wait options, this is not the cause of the problem. You seem to have correctly built the Bowtie2 index for the ref.fa reference but they remarkably (I have never seen this) have extensions bt2 instead of bt2l . Even if, indeed, as you suggest, renaming the *.bt2 files to *.bt2l could help, you of course are not expected to do so in aMeta, and to give you a better advice I need to check the ref.fa_BOWTIE2_BUILD.log file 🙂
*Thread Reply:* Okay, I just purged all modules and ran from scratch and now I have a different bowtie error! Here it is:
*Thread Reply:* ```Error in rule Bowtie2Index: jobid: 9 input: resources/ref.fa output: resources/ref.fa.1.bt2l, resources/ref.fa.2.bt2l, resources/ref.fa.3.bt2l, resources/ref.fa.4.bt2l, resources/ref.fa.rev.1.bt2l, resources/ref.fa.rev.2.bt2l log: resources/ref.faBOWTIE2BUILD.log (check log file(s) for error details) conda-env: /scratch/shreya23/aMetatest2/aMeta/.test/.snakemake/conda/6254db56e34ae5808d40831fc7518aca_ shell: bowtie2-build-l --threads 1 resources/ref.fa resources/ref.fa > resources/ref.faBOWTIE2BUILD.log 2>&1 (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile resources/ref.faBOWTIE2BUILD.log:
bowtie2buildl: unrecognized option '--threads' Bowtie 2 version 2.2.3 by Ben Langmead (langmea@cs.jhu.edu, http://www.cs.jhu.edu/~langmea|www.cs.jhu.edu/~langmea) Usage: bowtie2-build-l [options]* <reference_in> <bt2_index_base> reference_in comma-separated list of files with ref sequences bt2_index_base write bt2l data to files with this dir/basename Bowtie 2 indexes work only with v2 (not v1). Likewise for v1 indexes. * Options: -f reference files are Fasta (default) -c reference sequences given on cmd line (as <reference_in>) -a/--noauto disable automatic -p/--bmax/--dcv memory-fitting -p/--packed use packed strings internally; slower, less memory --bmax <int> max bucket sz for blockwise suffix-array builder --bmaxdivn <int> max bucket sz as divisor of ref len (default: 4) --dcv <int> diff-cover period for blockwise (default: 1024) --nodc disable diff-cover (algorithm becomes quadratic) -r/--noref don't build .3/.4 index files -3/--justref just build .3/.4 index files -o/--offrate <int> SA is sampled every 2^<int> BWT chars (default: 5) -t/--ftabchars <int> # of chars consumed in initial lookup (default: 10) --seed <int> seed for random number generator -q/--quiet verbose output (for debugging) -h/--help print detailed description of tool and its options --usage print this usage message --version print version information and quit
* Warning * 'bowtie2-build-l' was run directly. It is recommended that you run the wrapper script 'bowtie2-build' instead.
Error: Encountered internal Bowtie 2 exception (#1) Command: bowtie2-build-l --threads 1 resources/ref.fa resources/ref.fa ```
*Thread Reply:* I think I had bowtie loaded as a module separately when I got the last error, but here’s the log file for that run in case:
*Thread Reply:* @Shreya I think I understand the error. Strange that we did not notice it previously. Could you please open aMeta/workflow/rules/align.smk and add --large-index to this line:
bowtie2-build-l --threads {threads} {input.ref} {input.ref} > {log} 2>&1
so it should become something like this:
bowtie2-build-l --large-index --threads {threads} {input.ref} {input.ref} > {log} 2>&1
*Thread Reply:* If this fixes the error, we should quickly fix it in the main aMeta github repo. Apparently, as for now, the Bowtie2_Index rule builds small index, therefore produces *.bt2 files, while it should be a large index (for metagenomic projects) by default, hence *.bt2l files are expected by aMeta
*Thread Reply:* From the log-file you sent me, it did build the index correctly, just a small index, while it should build a large index (a large index is expected by aMeta), therefore you get this error. Strange that we never discovered this bug
*Thread Reply:* Ooh, let me give that a shot right now!
*Thread Reply:* Hmm, now I’m getting this:
```Error in rule Bowtie2Index: jobid: 9 input: resources/ref.fa output: resources/ref.fa.1.bt2l, resources/ref.fa.2.bt2l, resources/ref.fa.3.bt2l, resources/ref.fa.4.bt2l, resources/ref.fa.rev.1.bt2l, resources/ref.fa.rev.2.bt2l log: resources/ref.faBOWTIE2BUILD.log (check log file(s) for error details) conda-env: /scratch/shreya23/aMetatest2/aMeta/.test/.snakemake/conda/6254db56e34ae5808d40831fc7518aca_ shell: bowtie2-build-l --large-index --threads 1 resources/ref.fa resources/ref.fa > resources/ref.faBOWTIE2BUILD.log 2>&1 (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile resources/ref.faBOWTIE2BUILD.log:
bowtie2buildl: unrecognized option '--large-index'```
*Thread Reply:* Ok, sorry, this line should be correct:
bowtie2-build --large-index --threads {threads} {input.ref} {input.ref} > {log} 2>&1
*Thread Reply:* I’m deleting and re-cloning aMeta each time so it takes a few min
*Thread Reply:* as far as I understand bowtie2-build --large-index should be equivalent to bowtie2-build-l but for some reason it still builds a small index, which is strange, so hopefully bowtie2-build --large-index should be more stable
*Thread Reply:* @Shreya thank you for your patience, and yes, cleaning and re-installing is a good strategy in one encounters errors
*Thread Reply:* thank YOU for all your help!! Super excited to get this up and running 🙂
*Thread Reply:* bowtie2-build: unrecognized option '--threads'
Bowtie 2 version 2.2.3 by Ben Langmead (<a href="mailto:langmea@cs.jhu.edu">langmea@cs.jhu.edu</a>, <http://www.cs.jhu.edu/~langmea|www.cs.jhu.edu/~langmea>)
Is the threads option required?
*Thread Reply:* wait, Shreya, this is super-weird. --threads is a typical flag, I can't believe that bowtie2-build does not recognize it. Let me think a bit. Can you post the whole Bowtie2_Index rule from aMeta/workflow/rules/align.smk?
*Thread Reply:* Oh no I’m sorry! I’m certain I must have messed something up with my conda installing
rule Bowtie2_Index:
output:
expand(
f"{config['bowtie2_db']}{{ext}}",
ext=[
".1.bt2l",
".2.bt2l",
".3.bt2l",
".4.bt2l",
".rev.1.bt2l",
".rev.2.bt2l",
],
),
input:
ref=ancient(config["bowtie2_db"]),
conda:
"../envs/bowtie2.yaml"
envmodules:
**config["envmodules"]["bowtie2"],
threads: 1
log:
f"{config['bowtie2_db']}_BOWTIE2_BUILD.log",
shell:
"bowtie2-build --large-index --threads {threads} {input.ref} {input.ref} > {log} 2>&1"
*Thread Reply:* Also, I noticed that when I do conda list, I get this version of bowtie: bowtie2 2.5.1 py310ha0a81b8_2 bioconda , but the log file says 2.2.3
*Thread Reply:* @Shreya I think you are using some local version of Bowtie2, i.e. not the one installed togeter with aMeta. The latest Bowtie2 version is 2.5.2, this is what should be installed, while you seem to be using 2.2.3
*Thread Reply:* bizarre! It doesn’t appear that I have bowtie2 loaded as a module. Should I try installing and uninstalling it from base?
*Thread Reply:* @Shreya would you mind taking a screenshot of the very beginning of your testrun, I want to see a few first lines after you do:
cd .test
./runtest.sh -j 4

*Thread Reply:* Thank you, what do you see if you run now bowtie2-build -h ?

*Thread Reply:* 🤔 so it seems like a bowtie version issue
*Thread Reply:* Thank you so much for helping with this!!
*Thread Reply:* Also could you send me the full log-file located at .snakemake/log?
*Thread Reply:* @Shreya could you please do:
- conda activate /scratch/shreya23/aMetatest2/aMeta/.test/.snakemake/conda/6254db56e34ae5808d40831fc7518aca
- bowtie2-build -h
*Thread Reply:* Still Bowtie 2 version 2.2.3!
*Thread Reply:* I can see that the installation script is installing Bowtie2, but when you execute aMtea, an old version of Bowtie2 is run. So you either have this old Bowtie2 version somewhere in the base-environment (which still should not be a problem), or (more likely) your conda insists on installing the oldest Bowtie2 version
*Thread Reply:* Oh this is very strange! Yes, when I activate # packages in environment at /scratch/shreya23/aMetatest2/aMeta/.test/.snakemake/conda/6254db56e34ae5808d40831fc7518aca:, and then conda list, the installed Bowtie2 is 2.5.2
*Thread Reply:* Maybe I can go to base and try and upgrade the bowtie there and start from scratch
*Thread Reply:* Yes, please try conda install bowtie2=2.5.2
*Thread Reply:* Okay! I will do that and clean/reinstall aMeta and keep you posted… thank you!!!
*Thread Reply:* Or at least perhaps bowtie2=2.5.2 should be added instead of just bowtie2 aMeta/workflow/envs/bowtie2.yaml
*Thread Reply:* but on the other hand, you do seem to have the right bowtie2 version in your local bowtie2-environment /scratch/shreya23/aMetatest2/aMeta/.test/.snakemake/conda/6254db56e34ae5808d40831fc7518aca
*Thread Reply:* Okay so maybe bowtie2=2.5.2 will force the right version? I’ll try that next
*Thread Reply:* I would do both, do conda install bowtie2=2.5.2 in the base-environment (it seems to be heavily affecting aMeta for some reason, while it should not) and add bowtie2=2.5.2 to your aMeta/workflow/envs/bowtie2.yaml
*Thread Reply:* Okay, I have figured out a bit more!
If I don’t module load gccandmodule load python before running the test script, I get this error:
./runtest.sh: /home/shreya23/.local/bin/snakemake: /apps/software/gcc-6.2.0/python/3.6.0/bin/python3.6: bad interpreter: No such file or directory
*Thread Reply:* But if I load python beforehand, the pipeline starts running and breaks at the bowtie step. When I do “which bowtie2” it defaults to /apps/software/gcc-12.1.0/python/3.10.5/bin/bowtie2 , not the one packaged in aMeta. But if I unload python and do “which bowtie2" it gives me the aMeta version--but then I get the “bad interpreter” error. I’ve asked our sysadmin to update the installed bowtie2 to 2.5.2 so we’ll see if that helps!
*Thread Reply:* Hi @Shreya, thanks, I believe you should not load any modules at your HPC. Everything necessary should be delivered together with the aMeta installation. Including python. Also, this is a weird path /apps/software/gcc-12.1.0/python/3.10.5/bin/bowtie2. I do not understand how Bowtie2 can be related to python 🤔
*Thread Reply:* Hmmmm, okay. 🤔indeed! Thanks so much for the debugging — I will take this to the HPC admins and see if they can help sort things out. Will have to wait for after the Thanksgiving holiday but excited to get back to it soon! Thank you again Nikolay, I will keep you posted!
*Thread Reply:* @Shreya while you are waiting for the reply from the admins, I would recommend you to set aMeta up on your laptop. The testrun works fine on a laptop, and you will at least learn how a proper testrun output should looks like
*Thread Reply:* Your HPC seems to be special, all the errors you posted are new for me, although I have experience helping quite a few people working on different HPCs, but your base-env and module system interfering the aMeta environment seem extreme (therefore I even asked you in the beginning if you are sure you activated the aMeta environment, because everything looked like you did not 🙂 )
*Thread Reply:* Oh dear! I wonder why our HPC is strange. I did try to install it on my laptop but I got a segmentation fault after this bit 290724 packages in <https://conda.anaconda.org/conda-forge/osx-64> so I gave up on that and headed to the cluster!
*Thread Reply:* actually, installing aMeta on a laptop and running the testrun should not take longer than 5-10 min, I do it regularly (and on different laptops). Installing on an HPC can be generally more difficult compared to a laptop. So if you have time now, please go ahead, I will be available here for ~1 hour to assist you
*Thread Reply:* hmm, @Shreya is your laptop running Windows?
*Thread Reply:* nope, macbook air!
*Thread Reply:* Do you have a miniconda installed?
*Thread Reply:* yes, I have miniconda3 and have been able to install other packages before!
*Thread Reply:* Hmm, do you want to try again on your laptop now? Or you are busy with something else?
*Thread Reply:* I just tried it and got the same seg fault! I’m wondering if I need to update my conda installation or something
*Thread Reply:* I’m trying again with the conda option instead of mamba, just in case
*Thread Reply:* @Shreya what do you get if you do:
- mamba -h
- conda -h in your terminal (base-environment)
*Thread Reply:* ```(base) Shreyas-MacBook-Air:~ shreya$ mamba -h usage: mamba [-h] [-V] command ...
conda is a tool for managing and deploying applications, environments and packages.
Options:
positional arguments: command clean Remove unused packages and caches. config Modify configuration values in .condarc. This is modeled after the git config command. Writes to the user .condarc file (/Users/shreya/.condarc) by default. create Create a new conda environment from a list of specified packages. help Displays a list of available conda commands and their help strings. info Display information about current conda install. init Initialize conda for shell interaction. [Experimental] install Installs a list of packages into a specified conda environment. list List linked packages in a conda environment. package Low-level conda package utility. (EXPERIMENTAL) remove Remove a list of packages from a specified conda environment. uninstall Alias for conda remove. run Run an executable in a conda environment. [Experimental] search Search for packages and display associated information. The input is a MatchSpec, a query language for conda packages. See examples below. update Updates conda packages to the latest compatible version. upgrade Alias for conda update.
optional arguments: -h, --help Show this help message and exit. -V, --version Show the conda version number and exit.
conda commands available from other packages: env```
*Thread Reply:* ```(base) Shreyas-MacBook-Air:~ shreya$ conda -h usage: conda [-h] [-V] command ...
conda is a tool for managing and deploying applications, environments and packages.
Options:
positional arguments: command clean Remove unused packages and caches. config Modify configuration values in .condarc. This is modeled after the git config command. Writes to the user .condarc file (/Users/shreya/.condarc) by default. create Create a new conda environment from a list of specified packages. help Displays a list of available conda commands and their help strings. info Display information about current conda install. init Initialize conda for shell interaction. [Experimental] install Installs a list of packages into a specified conda environment. list List linked packages in a conda environment. package Low-level conda package utility. (EXPERIMENTAL) remove Remove a list of packages from a specified conda environment. uninstall Alias for conda remove. run Run an executable in a conda environment. [Experimental] search Search for packages and display associated information. The input is a MatchSpec, a query language for conda packages. See examples below. update Updates conda packages to the latest compatible version. upgrade Alias for conda update.
optional arguments: -h, --help Show this help message and exit. -V, --version Show the conda version number and exit.
conda commands available from other packages: env```
*Thread Reply:* Looks correct, your mamba are conda do not seem to be broken
*Thread Reply:* So instead of a segmentation fault when I try with conda, I get this now: ```(base) Shreyas-MacBook-Air:aMeta shreya$ conda env create -f workflow/envs/environment.yaml Collecting package metadata (repodata.json): done Solving environment: failed
ResolvePackageNotFound:
- megan```
*Thread Reply:* vs this for mamba:
```(base) Shreyas-MacBook-Air:aMeta shreya$ mamba env create -f workflow/envs/environment.yaml Getting conda-forge osx-64 Getting conda-forge noarch Getting bioconda osx-64 Getting bioconda noarch Getting pkgs/main osx-64 Getting pkgs/main noarch Getting pkgs/r osx-64 Getting pkgs/r noarch Getting r osx-64 Getting r noarch
Looking for: ['python >=3.7', 'snakemake-minimal >=5.18', 'mamba', 'pandas', 'biopython', 'fastqc', 'mapdamage2', 'multiqc', 'bowtie2', 'samtools', 'cutadapt', 'krakenuniq', 'krona', 'r-base', 'hops', 'seqtk', 'parallel', 'megan', 'r-pheatmap', 'pmdtools', 'pysam', 'pygments', 'jinja2', 'networkx', 'pygraphviz', 'imagemagick', 'graphviz', 'pandoc']
290724 packages in https://conda.anaconda.org/conda-forge/osx-64 9493 packages in https://conda.anaconda.org/r/noarch 5301 packages in https://conda.anaconda.org/r/osx-64 44225 packages in https://conda.anaconda.org/bioconda/osx-64 115641 packages in https://conda.anaconda.org/conda-forge/noarch 43793 packages in https://conda.anaconda.org/bioconda/noarch 31318 packages in https://repo.anaconda.com/pkgs/main/osx-64 9493 packages in https://repo.anaconda.com/pkgs/r/noarch 4910 packages in https://repo.anaconda.com/pkgs/main/noarch 5301 packages in https://repo.anaconda.com/pkgs/r/osx-64 Segmentation fault: 11```
*Thread Reply:* @Shreya do you have enough disk space on your laptop?

*Thread Reply:* @Shreya what do you get if you do:
conda install -c bioconda megan
*Thread Reply:* ```PackagesNotFoundError: The following packages are not available from current channels:
- megan```
*Thread Reply:* looks like megan is not available for mac in bioconda channel. It is certainly available for unix. Strange!
*Thread Reply:* I can only recommend you to install megain manually from here https://software-ab.cs.uni-tuebingen.de/download/megan6/welcome.html
*Thread Reply:* Unfortunately, I am going to have to head out soon — but I do have Megan installed already on my computer, so I will see what I can do to point the installation there.
*Thread Reply:* I believe you should use this file MEGANCommunitymacos625_6.dmg
*Thread Reply:* I see. I am very sorry about all the troubles. Installing software can be a pain. Please do not give up, we can have a zoom session and try to do the installation. Once it is installed, things will be much more stable
*Thread Reply:* No worries at all Nikolay, I’m sure the problem is somewhere at my end! I really appreciate your help with installing. I am very excited to get it up and running and have already promised a labmate I will screen her samples so I am certainly committed to getting it working!
*Thread Reply:* Good, thank you, and please do not hesitate to DM me to keep talking installation. I am available and will do my best to help you
*Thread Reply:* Thank you so much, I will certainly keep you posted!!
Hey all! Weird lab question - We're designing a homebrew capture to enrich our endogenous DNA content. We tested it on modern DNA and had no problems recovering the target sequences or weird behavior. However, when trying to capture our ancient libraries (which do have a different primer setup) we're getting some weird stuff: we are seeing amplification of our captured libraries (good), no amplification in our PCR negative (also good), but amplification in a "no probe control", where no baits were added to the libraries. The beads are streptavidin beads and the baits are biotinilated, so in theory only the baits should stick to the beads(?) Has anyone ran into something like this before? Perhaps an affinity of the beads for certain sequences, etc?
*Thread Reply:* This might be a naive question but have you tested whether your probes share a sequence similarity with spurious sequences that are not your intentional target? Blasting some of them against NCBI GenBank might give you an idea whether they might be able to anneal to DNA from other species, too. For me such a scenario seems more likely than that the beads themselves attract certain DNA molecules.
*Thread Reply:* Thanks Alex! We would blast the probes, but we've constructed them from random RAD fragments (so don't know what the sequences are). We do get some off target capture using this method (but it's negligible). Thanks for the suggestion - we were thinking the same think about the beads, but couldn't find much info on it.
*Thread Reply:* Do you have a taxonomic profile of your samples prior to enrichment?
*Thread Reply:* Without knowing the rough origin of your sequences that were used to design the probes, this is very hard to debug. But maybe there is someone here in the community who is more knowledgable about capture probes and binding specificity. Sorry!
*Thread Reply:* What temperatures were you using for the hybridization? Have you tested it at different temps?
*Thread Reply:* 60degC - We haven't played with the temps much, but writing this now that definitely feels like something we should do
*Thread Reply:* and @Alex Hübnerwe do! Appreciate the insights on things so far.
*Thread Reply:* We saw slight improvements at 65 degrees with our enrichments. Not sure if this will fix your issue but may be worth a try.

Hi! Has anyone has any problems when they try to build a BLAST database and it generates multiple header/index and option files e.g libraryblast.fna.00.nhr libraryblast.fna.00.nin libraryblast.fna.00.nog libraryblast.fna.00.nsq libraryblast.fna.01.nhr libraryblast.fna.01.nin libraryblast.fna.01.nog libraryblast.fna.01.nsq libraryblast.fna.02.nhr libraryblast.fna.02.nin libraryblast.fna.02.nog libraryblast.fna.02.nsq libraryblast.fna.03.nhr libraryblast.fna.03.nin library_blast.fna.03.nog

*Thread Reply:* I think this is normal for large databases, was there a particular problem you were encountering?
Hola! Does anyone have any sort of estimate or reference for the minimum amount of reads needed for proper ancient damage estimation? 📉
*Thread Reply:* Hola!
I did a mini simulation here: Mann, Allison E., James A. Fellows Yates, Zandra Fagernäs, Rita M. Austin, Elizabeth A. Nelson, and Courtney A. Hofman. 2020. “Do I Have Something in My Teeth? The Trouble with Genetic Analyses of Diet from Archaeological Dental Calculus.” Quaternary International: The Journal of the International Union for Quaternary Research, November. https://doi.org/10.1016/j.quaint.2020.11.019.

*Thread Reply:* Figure.... 4 or 5? Can't remember from my head
*Thread Reply:* Figure 2 (now it loads) so I was completely wrong 🤣
*Thread Reply:* But yes roughly what Nikolay said, but you can just about go down to 50
*Thread Reply:* But not reliabiy
*Thread Reply:* I've read this paper at least 20 times and I forgot that it was in there. I'm getting more and more thankful for this channel as my memory declines...
*Thread Reply:* But that's perfect. These references are exactly what I needed!
*Thread Reply:* > I've read this paper at least 20 times and I forgot that it was in there. I'm getting more and more thankful for this channel as my memory declines...

*Thread Reply:* Sowwy! Probably won't forget again 🥹 I keep using the potato and tomato example though! Can't even look at these vegetables in the same way anymore. 🍅🥔
@Aleksandra Laura Pach We recommend a minimum threshold of 100-200 in aMeta exactly for that reason, i.e. a convincing damage pattern is hard to get from fewer reads
Hello! I am performing metagenomic analysis on ancient DNA. I am currently following the eMeta but I am coming up to the point where I need to build a project-specific MALT database. Does anyone have any advice on this next step? I am trying to anticipate the amount of computing power and need and how to keep all of my files organized/backed up.
*Thread Reply:* Hi Jaime,
I would say it would be good to have at least 1 TB of RAM if not more. I would say try to make your database as comprehensive (i.e. large) as possible to improve your sensitivity. If you can get access to 3-4 TB of RAM, that would be ideal. But if not, you may have to reduce your database size.
You can use the krakenuniq-build to include the database or the NCBI Datasets command-line tools. I know aMeta uses krakenuniq so that may be the best approach.
I am not sure if you come across these papers below. But I think they may also help with what to think about when constructing a database and the amount of memory it would require. https://journals.asm.org/doi/full/10.1128/msystems.00080-18
https://peerj.com/articles/6594/
*Thread Reply:* Thank you so much, this is very helpful!
*Thread Reply:* Hey Jaime! Sterling is right, for the MALT database build step of aMeta, I would advise to use a node with 1TB RAM. And for the alignment step as well, by the way 😊

*Thread Reply:* You might be interested in taking a look at Struo2 (which works in conjunction with Kraken2/Bracken)! https://pubmed.ncbi.nlm.nih.gov/34616633/

*Thread Reply:* If your computational capacities are really limited I would recommend working (screening) target oriented on specific databases. For the detection of Metazoans I had really good results with the refsec mitochondrion database. It can also be used for plant detection. But if plants are the main target, it can also be beneficial to try screening with the refsec plastid database (also ncbi). When screening for microorganisms I had really good results implementing the SILVA SSU and LSU databases. It is quite easy to implement in the MALT workflow. One last thing that I can recommend when working with limited computational capacities is screening on sample at a time instead of a bulked operations. Much more work but it can prevent craching. Hope this can help :)
*Thread Reply:* Hi @Jaime Zolik, apologies for my very late reply! If you send me a DM with some details on the output of the KrakenUniq step (the one prior to the building of project-specific MALT DB in aMeta) I can advice you in details about recommended resources. So far it is hard to guess since I do not know how many microbes were detected in the first (KrakenUniq ) step
Hey everyone! I have a Kraken database question. Does anyone have experience reducing the built database to some maxdbsize and if so, did you notice some loss of sensitivity?
*Thread Reply:* I've not directly experienced it myself, but it makes sense -IIRC you're reducing the number of kmers by down sampling to try and keep the most informative ones, but the most informative ones aren't the only ones that would also be valid.
If you remove any amountreference k-msrs you will reduce the chances of getting a hit
@Maxime Borry probably knows better
*Thread Reply:* I can attest to what James is saying. With Kraken uniq and kmer set at 31, we were able to still run malt build. We noticed a considerable reduction in the number of species. It is my understanding that a lot of this is discussed in the aMeta paper and they have a lot of the data to show how sensitivity and specificity is impacted by changing the database size.
*Thread Reply:* Hi @Sarah Johnson, reducing database size will certainly lead to reduced sensitivity (and indirectly specificity). We discuss this here https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03083-9, please see the section "Effect of database size". Regarding your particular question about reducing Kraken DB size with maxdbsize, if I recall correctly (perhaps it was in KrakenUniq and not Kraken2) this option was broken long time ago. I remember I tried to use this option, and was still running out of memory when building a database. But perhaps they have already fixed it, or I remember incorrectly, so I would encourage you to try and let us know how it went, I would be curious to know your experience. Still, I guess, you would not like to reduce you DB size because you do not want to sacrifice analysis accuracy, but you probably have to do it because you do not have enough RAM. This is unfortunately a typical problem in this field: the balance between the accuracy and resources, there is no straightforward solution here 😞
*Thread Reply:* Thank you all for your responses. Yes, I’m more concerned with resources. The HPC at my university has the resources but it is shared across the university so I’m trying to sacrifice as little specificity and sensitivity as possible while optimizing resource utilization. I think I might try a small experiment looking at that if I have time and will update as I have more information. Kraken1 has the ability to shrink databases but I’m not sure if one built with kraken2 would be shrink-able.
*Thread Reply:* It's definitely working for kraken2, that's how they do it for https://benlangmead.github.io/aws-indexes/k2
*Thread Reply:* Thanks @Maxime Borry! @Sarah Johnson I would suggest that perhaps an intelligent way of reducing database size would be excluding redundancy and keeping representative organisms per e.g. genus or family. This however needs a lot of time and manual work. But if you have time and energy, this way could result in a dramatic database reduction without (presumably) loosing much accuracy
*Thread Reply:* @Nikolay Oskolkov I had the same thought. I wrote a little script to do just that. We’ll see. Im going to try shrinking and/or building a reduced db and will let you know.

Hello! I have a question re: phylogenies. I found an interesting bacterial species when running MALT (or actually the aMeta workflow). I want to find out what strain of the species the reads belong to but the coverage is low. My idea was to create a phylogeny using a few known strains and see where my reads end up in the tree. However, I've failed massively so far so I'm turning to you for some input 🙂 Has anyone here done something similar, or read about something similar in a paper somewhere? What I have done so far is downloading different strains, cat them together with the reference genome used in MALT and my sequence data and tried to create a consensus alignment. I think the fact that the sequence data is so fragmented makes it difficult to create the consensus because I keep running out of memory on our cluster.
*Thread Reply:* I can add that I tried a run with fewer samples (meaning I forgot the outgroup...) and then it worked fine, creating a consensus with muscle and then the tree with BEAST. So I think I'm not way out of line with this idea but obviously something is not right
*Thread Reply:* Oh also, I realise selecting a few genes to focus on could be an idea but I'm afraid I won't have coverage of full genes in my sequence data
*Thread Reply:* Hi Nora, i once used pathPhynder to place low coverage genomes into a given phylogeny. https://doi.org/10.1093/molbev/msac017 not sure what the minimum coverage would though (always wanted to test that). it’s quite easy to use.
*Thread Reply:* I second pathPhynder for this. It has also been applied to very low coverage situations (ie. ancient environmental DNA)
*Thread Reply:* Or EPA-ng, non? Just include your low-coverage genome in your alignment (it will be full of N’s), build the tree with the good genomes only, and then place the low-coverage genome in that tree
*Thread Reply:* To do a @Antonio Fernandez-Guerra ;):
nf-core has a pipeline for EPA, maybe that would be helpful?

Hey everyone, What’s your experience with haplotype phasing/strain deconvolution for your short metagenomic read mapping to bacterial genomes ? (ie: getting the consensus sequence/haplotype of each strain when you have multiple strains mapping to a species reference genome).
*Thread Reply:* Hi Maxime, inStrain is currently the best SNP-based approach to do this: https://www.nature.com/articles/s41587-020-00797-0 SynTracker is a newer tool that does the same thing, but based on synteny (making it insensitive to SNPs): https://github.com/leylabmpi/SynTracker
*Thread Reply:* But aDNA with our very short reads is kind of excluded for inStrain > For each read pair aligned to the reference genome (de novo assembled from the same sample or a genome from another source) the mapQ score, average nucleotide identity (ANI) of the pair to the reference genome and the insert size between aligned reads are calculated. Read pairs that do not pass adjustable quality cutoffs are removed, as are all unpaired reads. The exclusive use of pairs doubles the number of bases used to calculate the read ANI and mapQ score, increasing their accuracy and substantially increasing the span of genome analyzed.
*Thread Reply:* Most of the time, the aDNA fragments are shorter than the reads, so paired reads are merged (“negative” insert size) because the forward and reverse reads actually overlap
*Thread Reply:* Syntracker requires metagenomic assemblies 😞
*Thread Reply:* Oh, right, sorry I am new here 😅 If you haven’t already, I’d recommend reaching out to Matt Olm on inStrain’s Git page! He’s super responsive; would likely have ideas
*Thread Reply:* Good idea, thanks @Liam Fitzstevens 🙂
*Thread Reply:* Assuming you have a mix of two haplotypes, you could try the method presented in https://www.nature.com/articles/s42003-021-01710-4 to deconvolute the two sequences.
*Thread Reply:* Alternatively there is Kallisto, developed by @Benjamin Vernot’s group. This does not pull out the consensus sequences but can give an indication of the relative abundance of different haplotypes (and will work with >2 haplotypes).
*Thread Reply:* Thanks @Pete Heintzman You mean dividing your variants into high and low frequency set ?

*Thread Reply:* Hi! Regarding this question I also tried to use Instrain profile with ancient DNA (short reads). I collapsed the reads before mapping it against the genome reference and I have a bam file that I give as input to instrain. I used the flag --pairingfilter nondiscordant (non_discordant = Keep all paired reads and singleton reads that map to a single scaffold) to keep all reads in the analysis. I am new to this kind of analysis, so maybe it is not the best approach to bypass the problem with the short fragments.
*Thread Reply:* Kallisto wouldn't give you the consensus sequences, but if you had a collection of strain references it might be pretty good for picking out the best mixture
*Thread Reply:* Lots of caveats, though. Happy to chat when I'm back.
And related question: do you have any recommendation for ploidy estimation (ie: number of strains) for aDNA short reads ?
*Thread Reply:* Variation graph would be one solution, but only for well studied species, where there are already all the variants in the reference genomes variation graph.
*Thread Reply:* depending on what genomes you're working with, you could try treating strain-specific regions as a "chromosome" and look at coverage variation between chromosomes? this only really works if you have variable gene content though. you could also try using heterozygosity in different regions as a proxy for this? I'm not sure how this would be impacted by e.g. contamination though, but might be worth trying
Hi all! Has anyone tried to use the consensus_aDNA.py script from cmseq? I have a rescaled bam file (I used mapdamage2 to rescale it) that contains reads from an ancient bacteria aligned against the genome reference. This is the command that I used:
consensusaDNA.py --mincov 5 --minqual 30 -r genomeref.fasta --posspecificprobtab StatsoutMCMCcorrectprob.csv --posdamageprobthrsh 0.95 bamrmdupsort.rescaled.bam
And this is the error:
Traceback (most recent call last): File "/softwares/condaenvs/metagenomics3/bin/consensusaDNA.py", line 10, in <module> sys.exit(consensusfromfile()) File "/softwares/condaenvs/metagenomics3/lib/python3.7/site-packages/cmseq/consensusaDNA.py", line 265, in consensusfromfile trimReads=None,postdamageprob=posprobthrsh,posprobdb=posstatsdb, refseqidx=RefSeqidx) File "/softwares/condaenvs/metagenomics3/lib/python3.7/site-packages/cmseq/consensusaDNA.py", line 98, in referencefreeconsensus consensuspositions[pileupcolumn] = consensusrule(dict((k,v) for k,v in positiondata['basefreq'].items() if k != 'N')) File "/softwares/condaenvs/metagenomics3/lib/python3.7/site-packages/cmseq/cmseq.py", line 206, in majorityrule freqarray= dataarray['basefreq'] KeyError: 'basefreq'
Has anyone dealt with this error?
Thank you in advance!
*Thread Reply:* You’re in luck, the author of this code is in this workspace 🙂 @Kun Huang
*Thread Reply:* If you decide to use something else than cmseq, I’d recommend calling your variants (using freebayes for example) from your mapdamage rescaled bam file, and then filter on variant support and/or quality (bcftools)
*Thread Reply:* bam -> freebayes -> bcftools view -> bcftools consensus
*Thread Reply:* Thank you very much for your help! 🙂
*Thread Reply:* And what if I use angsd to create the consensus sequence (using as input the mapdamage rescaled bam) and then I merged all the fasta from the different individuals and use snp-sites to have a multifasta only with the snps? Is it a good idea or it is not a good approach for ancient data?
*Thread Reply:* Any variant caller should work (if using sensible parameters). My go to caller is freebayes 🙂
Hi all, I have a question about MultiVcfanalyzer: https://github.com/alexherbig/MultiVCFAnalyzer. I am basically trying to call SNPs and to get a full alignment from several bams. For that, I used Eager 2.5 for all the mapping, filtering, and genotyping steps. Unfortunately at the snp table generation step I always get an error message from MultiVCFAnalyzer. I replicate the issue with versions 0.85.1 and 0.85.2 of MultiVCFAnalyzer. I always get this error message which I really do not understand: MultiVCFAnalyzer - 0.85.2 by Alexander Herbig
No positions to exclude provided! All positions will be used! Now processing 1/182: 0ad1bd59442cf28fa247368a0a9603 Illegal arguments in function getAmbiguousBase: R, G Illegal arguments in function getAmbiguousBase: M, A Illegal arguments in function getAmbiguousBase: Y, T Illegal arguments in function getAmbiguousBase: S, C Illegal arguments in function getAmbiguousBase: W, A Illegal arguments in function getAmbiguousBase: K, G Illegal arguments in function getAmbiguousBase: K, T Illegal arguments in function getAmbiguousBase: S, C Illegal arguments in function getAmbiguousBase: K, G Illegal arguments in function getAmbiguousBase: Y, T 500000 positions processed. Illegal arguments in function getAmbiguousBase: W, A Illegal arguments in function getAmbiguousBase: K, T Illegal arguments in function getAmbiguousBase: W, A Illegal arguments in function getAmbiguousBase: R, A Illegal arguments in function getAmbiguousBase: W, T 1000000 positions processed. Illegal arguments in function getAmbiguousBase: S, C Exception in thread "main" java.lang.ArrayIndexOutOfBoundsException: 1178035 at MultiVCFAnalyzer.main(MultiVCFAnalyzer.java:337) . I was wondering if some other people got the same errors and how did you manage to fix them? And if not, what are you using instead of MultiVCFAnalyzer (especially a tool which have "Minimal allele frequency for homozygous call" and "Minimal allele frequency for heterozygous call" options). Thanks in advance 🦠 !!
*Thread Reply:* Hi, I have seen similar errors but not that specific one. Which genotyper are you using?
*Thread Reply:* Which parameters are you using for the genotyping step? I think I’ve seen something like this when the vcfs are not exactly how multivcfanalyzer expects them
*Thread Reply:* Which reference genome are you using (if you can say)?
MultiVCFAnalyzer doesn't work with multi-chromosome references...
*Thread Reply:* But honestly the best way will be to email Alex Herbig... 😅 the error messages are renowned for being useless
*Thread Reply:* --rungenotyping --genotypingtool 'ug' --gatkploidy 2 --gatkugoutmode 'EMITALLSITES' --gatkuggenotype_model 'SNP'
*Thread Reply:* It is a bacteria reference genome with 2 chromosomes, so it could because of that
*Thread Reply:* Ok, that looks right. Yeah, the multiple chromosome is a thing
*Thread Reply:* Ok, i might try again with only one chromosome to see if the issue is coming from that
*Thread Reply:* Update, it is working when i divided the work by chromosome, thnaks a lot!
*Thread Reply:* > Ok, that looks right. Yeah, the multiple chromosome is a thing @Zoé Pochon (saw your reaction), MultiVCFAnalyzer was apparently never meant to be proper software, it was sort of a 'throwaway script' which kept being used 😅
Aida and I were talking about it the other day and realised probably just can do the same thing (more reliability) with a wrapper around bcftools consensus if we come up with the correct filtering conditions...
*Thread Reply:* (although won't get the SNP table etc, but still)
*Thread Reply:* Yes, i was thinking about using a handmade script, but I do not think that any other software allows you to play with the heterozygosity, is it?
*Thread Reply:* As long as you use a variant caller that allows reporting of multiple alleles, bcftools should allow (if you work out the filtering conditions,). You run that on each of your genomes and then cat the resulting fasta, you've pretty much got it... In theory...
I wouldn't make a whole script to parse the vcf etc when bcftools probably does it really well already

Hi everyone! This is my first post in this group so hello and nice to meet you all virtually! I have many no-stupid-questions questions! I’m looking for some clarification when it comes to using Kraken2 and Ganon. I’m going to ask my Ganon questions later. Today is about the beast, kraken.
The PDF will have most of this along with the tables and stuff that didn’t want to copy over well.
Just as some background, we are trying to use read classification tools for multiple aDNA projects - so keep in mind, everything is short and damaged. We’ve built one database that has fish species (along with human and Unive_Core) in the hopes of being able to use it to identify samples to the species level for fish bones that are morphologically hard to distinguish in archeological contexts. We’ve built a much larger database with ALL the RefSeq genomes (limited to 3 assemblies per species). We were hoping to use this RefSeq db for sediment and coprolite projects - some examples of what we are trying to find include environment/host microbial communities, host diet, and for some of the sediment stuff we want to see if we can identify animal community too.
For Kraken2, does anyone have experience building Kraken2 databases with different kmer and minimizer parameters? We noticed with some of our stuff, shorter reads weren’t being classified when using the default parameters, so we wanted to try shorter kmer lengths than the default, and they just don’t seem to be classifying reads as well, especially to lower taxonomic ranks like genus and species.
Here is some information on the build parameters used for 2 builds with the same references as input: --kmer-len 25 --minimizer-len 21 --minimizer-spaces 4 • 2,552,952,570 total kmers • 14.5 GB Estimated hash table requirement --kmer-len 35 --minimizer-len 31 --minimizer-spaces 7 • 9,600,970,913 total kmers • 54 GB Estimated hash table requirement Question1: Why does the 35 kmer db have so many more total kmers? I’ve always thought that if you decrease kmer length, you’d have more kmers and larger database files, but I’m seeing the opposite, please help me understand!
Moving on, I tested the two dbs by running one sample through each db with the same classification parameters (minimum hit 4, confidence 0.10):
Question2: Does anyone have recommendations for minimum hit groups and confidence thresholds for aDNA reads? And can you tell me why those are your recs? I’ve done some tests on simulated data (with damage) using different parameters, but I’d love your input too. • I know confidence thresholds can help balance between filtering out spurious matches and allowing for potential errors due to ancient damage. So what confidence thresholds are aDNA ppl using (Q2.a1)? And would you all recommend using different confidence thresholds when trying to identify microbial community vs animal species (like the classifying reads to fish species/host diet stuff)? (Q2.a2) • I’ve been thinking lately that maybe a minimum hit of 4 might be too high for shorter reads and lowering it could increase the likelihood of classifying shorter reads. Are people just using the default (i think its 2)? (Q2.b1) ◦ Bonus points if someone can reassure me that I’m thinking of minimum hit groups correctly (Q2.b2): ▪︎ see PDF for options/example • I thought it was option1 when I was first testing these parameters, but after reading the brief description of it in the kraken2 readme a few months back, I think it is option2 now (love that I probably misinterpreted this initially). See PDF for tables with results from the test of the two dbs. I ran one sample through each db with the same classification parameters (minimum hit 4, confidence 0.10): • It initially seems like the 25 kmer db is classifying more reads, which is great. But it seems like a lot of those reads are being classified to higher taxonomic levels that aren’t as informative for our research qs like order, suborder, and family. And when you get to the species level, it seems like the 25kmer db decreases in precision and sensitivity (still don’t fully understand how these 2 benchmarking terms are different in total honesty). It’s top 2 species hits were to species that aren’t even in the correct genus! • Q3: For people who have been using shorter kmer lengths, are you noticing this pattern too? Have you found a way to mitigate it whether that is using different build or classification parameters? Because theoretically it would be nice to have a shorter kmer length to be able to classify shorter reads, but at the same time I don’t really want to sacrifice precision and sensitivity. Looking forward to your input and expertise!!!

*Thread Reply:* Hi @Karissa Hughes, a lot of questions, ha-ha 🙂 I will come to some of them a bit later (do not have much time right now), but your question 1 is more or less obvious, so I will start with this one. The numbers of k-mers that you mention in your post are related to the numbers of possible combinations of constructing 25- and 35-character words out of 4 letters (nucleotides). For example, you can construct 4^2=16 possible 2-character words out of 4 letters (A, C, T, G), i.e. you can pick 16 different pairs of letters out of 4 possible letters. If you want to construct 5-character long words using only 4 letters, you can build 4^5=1024 such words. And so on. So the longer words you would like to build out of a fixed number of letters, the larger "dictionary" you will get, so the database size will increase if one wants to achieve higher analysis specificity and use longer k-mers. Now, in your case 4^25 and 4^35 are much greater numbers than the ones you reported in your post. I believe this is because not all possible k-mers are used in the database in the end. Probably some theoretically possible k-mers were never detected in your reference genomes, this is one reason. Another reason is that, for example in text analysis, people usually severely prune their dictionaries and keep only most common words, so ultra-rare k-mers (e.g. seen in just one reference) were probably dropped from your final database. Hope this helps for the beginning, and as I said I will try to come back to your other questions a bit later when I have more time
*Thread Reply:* Q2: we need to find an alternative to Slack and its limitation to 90 days of message archiving on a free plan, because this has been answered multiple times already 😞 But TLDR: using a combination of number of reads, and duplication rate is a good starting point. In Kraken2 it’s slightly different , but as food for thoughts, this is a starting point https://maximeborry.com/post/kraken-uniq/
*Thread Reply:* Q3: This behaviour is expected. Shorter k-mers are less specific, meaning that they will be found more often than longer ones. So you would get more classified reads, especially if you have sequences in your dataset that are not represented directly in your reference database. However, because these are less specific, they will be found in more reference sequences than longer k-mer. And the more you find a given k-mer in different refs, the more the LCA will send it back to less precise taxonomic ranks.
*Thread Reply:* Given a 4 base alphabet (A,T,G,C), the probability of finding a given k-mer of k is (1/4)^k
*Thread Reply:* This one also seems to be a nice way of post processing kraken2 results https://www.biorxiv.org/content/10.1101/2024.02.02.578701 haven’t tried it though https://github.com/cdiener/architeuthis
*Thread Reply:* Thank you all for your replies so far! I appreciate it! I'll take a look at all the material you've shared before I ask any follow up qs

Hello everyone! It's my first post here, as I'm new to the community, and I have questions about the worflow eager. I'm currently a MSc student doing an internship and I'm working on ancient data of human dental calculus. I built my tsv file like this : SampleName LibraryID Lane ColourChemistry SeqType Organism Strandedness UDGTreatment R1 R2 BAM ROU2014 lr41 1 2 SE Human double none /work/project/GenIn/ROUSILLE/data/concatsamples/ROU2014lr41.fastq.gz NA NA ROU2030A lr46 1 2 SE Human double none /work/project/GenIn/ROUSILLE/data/concatsamples/ROU2030Alr46.fastq.gz NA NA ROU2031 lr48 1 2 SE Human double none /work/project/GenIn/ROUSILLE/data/concatsamples/ROU2031lr48.fastq.gz NA NA
I specified the SeqType as SE because my reads have been trimmed and collapsed, even though they are originally paired-end. I skipped adapterRemoval and followed the tutorial on metagenomics. For the MALT database I used the script "099-refseqgenomesbacteriaarchaeahomocompletechromosomescaffoldwalkthrough_20181122.Rmd" from the article "The evolution and changing ecology of the African hominid oral microbiome" from 2021.
I'm wondering if I specified correctly the SeqType and if I have to put that they were originally paired end what should I put in the R1 and R2 columns?
I ran the workflow on the tsv file and it processes correctly until the maltextract step where I get the error "Danger empty keys in File" when eager try to read my rma6 files. I tried to visualize one of them in MEGAN but I'm not used to this software so I can't see any issues with my rma file.
If you could help me with those steps, I would be very glad. Thank you for the opportunity to exchange with the community.
Best regards, Mathias CAIRE
*Thread Reply:* Hi @Mathias Caire! For nf-core/eager specific questions we have a dedicated pipeline slack channel you can access via the nf-core slack: https://nf-co.re/join
However I'm in both so can answer here🙂
For the SeqType, you're correct there to set as SE if they are already collapsed 👍
For the maltextract error, can you supply your full eager command? If possible please put in a code block (put triple backticks befroe and after the command)
*Thread Reply:* maltextract can be pretty wonky -- if you get the rma6 files to your local machine and open them with MEGAN you could see if the issue is with the general MALT step or with maltextract specifically
*Thread Reply:* And for the code block: https://commonmark.org/help/tutorial/09-code.html#:~:text=To%20create%20a%20code%20block,and%20below%20the%20code%20block.&text=A%20code%20block%20or%20span,4%20spaces%20or%20one%20tab.
*Thread Reply:* Thank you, I'm relieved that I specified correctly the SeqType. The eager command of maltExtract is: ```#!/bin/bash -euo pipefail MaltExtract -Xmx1024g -t coregenera-anthropoidshominidspanhomo-20180131.txt -i lr41.unmapped.rma6 lr46.unmapped.rma6 lr48.unmapped.rma6 -o results/ -r hops -p 64 -f defanc -a 0.01 --minPI 85.0 --destackingOff
postprocessing.AMPS.r -r results/ -m defanc -t 64 -n coregenera-anthropoidshominidspanhomo-20180131.txt -j``` Do you need more informations about how I ran the workflow? I also registered on the nf-core slack, do we continue the conversation there?
*Thread Reply:* Let's keep it in one place for now, and stay here but in the future we can switch
*Thread Reply:* Can you head the core_genera_..... file?
*Thread Reply:* I simply took the one from the tutorial and the whole file is :
Streptococcus
Tannerella
Porphyromona
*Thread Reply:* What MALT database did you use/
*Thread Reply:* I built it thanks to your script : "099-refseqgenomesbacteriaarchaeahomocompletechromosomescaffoldwalkthrough_20181122.Rmd" So it's a RefSeq database of bacteria archaea and homo sapiens with 56.104 genomes in it, I indexed it with malt-buid
*Thread Reply:* What version of MALT?
*Thread Reply:* (NGL I'm extremely impressed that you've been able to understand that horrific github mess 😆 )
*Thread Reply:* Hmm, that could be the problem
*Thread Reply:* What was the malt-build command?
*Thread Reply:* You likely will need to re-build the database but with the same or older verison of MALT as I used...
*Thread Reply:* I work on the genotoul cluster so it's a SLURM script : ```#!/bin/bash
SBATCH -p workq
SBATCH -t 01-00:00:00
SBATCH --cpus-per-task 64
SBATCH --mem=2000G
SBATCH -J "malt_indexing"
SBATCH --output=slurmoutput/indexing%A.out
Warning! Default threads number:64 (don't forget to adjust -t options of malt command and --cpus-per-task for Slurm).
Load modules
module load bioinfo/MALT/0.6.2
malt-build -t 64 -i references/. -d index --sequenceType DNA -J-Xmx2000g```
*Thread Reply:* No --mapdb? or --acc2tax?
*Thread Reply:* That may be your problem in this case
*Thread Reply:* If I'm honnest, I am not familiar with those options, I will investigate this thank you!
*Thread Reply:* Hey, me again! I do need to specify a file mapping RefSeq identifiers to taxon ids. Upon searching, I found tables that link accession numbers to taxon ids at
<https://ftp.ncbi.nih.gov/pub/taxonomy/accession2taxid/>
However, it seems they are using GenBank accession numbers. Do you happen to know where I could find the equivalent for RefSeq accessions?
I have the taxid of the assemblies I used to build the MALT database, so I'm currently creating a custom table, but I'm unsure about which format to use.
Thank you very much for your assistance.
*Thread Reply:* I think the genbank and refseq accessions are in the same file
*Thread Reply:* nucl_wgs.accession2taxid.gz TaxID mapping for live nucleotide sequence records of type WGS or TSA.
nucl_gb.accession2taxid.gz TaxID mapping for live nucleotide sequence records that are not WGS or TSA
*Thread Reply:* From the acc2tax README
*Thread Reply:* It's not raw reads (WGS) as refseq should be assemblies so presumably that counts as the second?
*Thread Reply:* Refseq is just a 'cleaned up' genbank
*Thread Reply:* Thank you for the time you're dedicating to me, it helps me a lot. I will try this.
*Thread Reply:* You're welcome! I'll be AFK from tonight for a couple of days as we're flying to Australia, but if you have more questions just make a new post on this channel and someone else can help you :)
I goofed real bad today and started a double-stranded partial-UDG lib prep (https://pubmed.ncbi.nlm.nih.gov/25487342/) with only enough T4 PNK for half the samples. After the UGI incubation, I put half in the freezer. Does anyone know if they are stable at this point? Can I continue with blunt end repair in a week or so when the T4 PNK arrives?
*Thread Reply:* In theory, this should be fine as there is no immediate need to end repair the partial-UDG treated DNA. Good luck!
*Thread Reply:* Awesome. This is what I was hoping! Thank you!

Hi there, I am trying to index my bacterial database with MALT and I consistently get java errors. I increased the malt memory in malt-build.vmoptions and also ran “set JAVAOPTS=%JAVAOPTS% -Xms1024m -Xmx1024m” I am still getting “java.lang.OutOfMemoryError: Java heap space” Does anyone know how to solve it or where the problem may be?
*Thread Reply:* For the VM options there you've only set a gigabyte of memory, is that correct?
*Thread Reply:* Also I think it's not good to set both of those parameters in Java (I can't remember exactly, Java memory management is a mystery for many people)
*Thread Reply:* so in the past we'vce set it like:
*Thread Reply:* malt-run J-Xmx1800G <....>
*Thread Reply:* That sets for to 1.8TB
*Thread Reply:* I set it to 64GB and a few other values just to see what works, if anything, and since it did not work I also changed the other parameter. I will try to run it again using your suggestion
*Thread Reply:* How many bacterial genomes
*Thread Reply:* I will know tomorrow and get back to u!
*Thread Reply:* Ok! Because 64GB seems quite small, we normally run for e.g. >1000 genomes a couple of hundred of GB
*Thread Reply:* okay then I guess it was too small. I am using all complete genomes from the ncbi
*Thread Reply:* Ooooooh yeah you'll need more than that 😅 you can also reduce the step size a bit to reduce the memory requirements. In 2017 we needed 300GB for a similar database
*Thread Reply:* what do you mean by step size
*Thread Reply:* From the malt-build documentation:
*Thread Reply:* It's the size of the seed offset. You lose detection/alignment sensitivity slightly, but it's minor if you set to a step size of 4 or 8 (any more of that you get a big drop off)
*Thread Reply:* I will play around with these on Thursday when I try to rerun the indexing.
*Thread Reply:* I increased the memory to 1200GB and I get this.. “Opening file: table0.db Allocating: 1010 GB java.lang.OutOfMemoryError: Java heap space” and “Exception in thread “Sync Timer Thread” java.lang.SecurityException: Could not lock User prefs. Lock file access denied.” … T.T
*Thread Reply:* There is some progress tho because the table0.db file is not empty. But that’s it
*Thread Reply:* With the -J parameters?
*Thread Reply:* But your might need to keep going higher... Did you try dropping the step size too?
*Thread Reply:* It complained about the -J command, maybe I wrote something wrong. I went higher and it is running. Complained “Exception in thread “Timer-0” but despite that it keeps running.
*Thread Reply:* Ok I think you can ignore the time-0... but let's see if it completes...
*Thread Reply:* Update: increasing memory worked and I screened one sample. Now I am trying other samples and it does not work.. It just stops midway. I get the output file, but I think it is incomplete. I get no error at all. I increased the memory further and nothing, I will try to play around with the step size as you suggested
*Thread Reply:* Ah step option is just for indexing xD I do not know why the screening does not work for all samples. Maybe not enough memory..
*Thread Reply:* Are you submitting to a HPC?

Hi! I need a little orientation with MALT anyone here had been working with it? The issue is: I am using the version 0.6.2 of MALT and in my command I need to use "--classify Taxonomy", but it is not working because it said it does not exist, in the terminal, but it exist in the manual for that version. So I can run it with a lower version as 0.4.0 but not with the 5 or the last one as 6.2. This variable "--classify Taxonomy" is important in my command because with out it does not make the correct assignation. Also I have change it just putting -c Taxonomy and in the complete way. I'll be glad to hear some advice.
*Thread Reply:* Don't trust the manual, it's very weirdly out of date - check the help message. Also don't use the mapdb it mentions, it's broken. Use the deprecated acc2tax flag (it still works) with the ncbi acc2tax map
*Thread Reply:* I hava the same error with acc2tax. And my command is malt-build -i Virusallseq.fasta.gz -d indexpruebatax -s DNA --acc2tax NCBI.txt Also I read the samen problem in MEGAN community but no one answer, I just replay it to know if the person could fix it.

*Thread Reply:* Was this one, dated last year https://megan.cs.uni-tuebingen.de/t/malt-build-assign-wrong-taxonid-on-some-reference-genomes/2169/2

*Thread Reply:* Hmm ok, I definitely used it a couple of weeks ago, when I get to my laptop I'll share with you the command I used...
*Thread Reply:* Ahh wait, try the short version of the parameter!
*Thread Reply:* Like -a2t or something!
*Thread Reply:* I think that was another bug...
*Thread Reply:* I use -a2t and know is running I'll wait to the result
*Thread Reply:* It works! thanks 😄

*Thread Reply:* Hi James, I have the exact same problem as this post (some phage such as S. mitis phage (AY007505.3) mis-assigned into S.mitis (taxid 28037)): https://megan.cs.uni-tuebingen.de/t/malt-build-assign-wrong-taxonid-on-some-reference-genomes/2169/2 I am using MALT/0.6.2, any idea to solve it? Thanks !
*Thread Reply:* Unfortunately, no I don't know. I realise now I forgot to say in my first reply: I don't really recommend MALT anymore because there seem to be a lot of bugs... (Sorry about that @Ania TSL
*Thread Reply:* It might be worth also replying saying it's a problem, the more people saying they have this issue, also in later versions of malt, the more likely it would be fixed
*Thread Reply:* Some recommended HOPS as a kind of substitution of MALT but I need to learn a bit more of it, also right now it is working so maybe if I have more problems I'll check it with more details.
*Thread Reply:* HOPS is a pipeline that has MALT in it
*Thread Reply:* You can try using the last reported version of the malt which I think was either 0.38 or 0.40
*Thread Reply:* @Yuejiao Huang use -a2t with the accession numbers for the NCBI and it works
*Thread Reply:* Oh interesting! @Ania TSL maybe you can post that on the forum thread?
*Thread Reply:* Yes I did it.

*Thread Reply:* Could you share your final version of command if you solved this problem?:) I am also trying to build a malt custom database🥹.
*Thread Reply:* Hi, sorry for my late response I did not notice the message until now, are you still working on it? The final version is: malt-build -i ~Virusallseq.fasta.gz~ -d indexpruebatax -s DNA -a2t ~NCBI.txt~ Let me know if it works for you @Wenqin Yu
*Thread Reply:* Thanks for your reply, Alitery! I’ve tried MALT for a long time, but it always returned a memory space error unless I provided a database that only includes viruses. Then I tried HAYSTAC, and it seems to be working so far. Thanks you again!!! :)
*Thread Reply:* I use #$ -l vf=10G
$ -pe openmp 20
export ompnumthreads=24 You need to be sure that you have enough space, If you want we could have a meet by zoom to check specific points. I am working with a db of virus too.
*Thread Reply:* It doesn't seem too resource-intensive! Thanks for your sharing, Ania! I'd like to hear more about your settings at your convenience.
Hi all! I am trying to run pydamage on my assembled ancient metagenomic data, however, when I do the filter step (for pydamage analyse I used the flag -w 30 and for the filter -t 0.67) I end up with few contigs (62 out of 173459). I already analyzed this sample to be sure that it contains ancient sequences, so I was not expecting this result. Does anyone have this same problem with assembled ancient metagenomic data? I am attaching a plot with the result for one of the contigs that was removed after the filter step.

*Thread Reply:* The pale pink line of the actual frequency looks a bit funky, it implies none of your reads have a C-T on the first position, it's that to be expected? We often see that when there is a artefact left on the read
Otherwise @Alex Hübner @Maxime Borry can hopefully advise
*Thread Reply:* Yes, this looks rather odd. Would you expect this from building your libraries? How did you assemble the contigs and align the data?
*Thread Reply:* I used metaSPAdes with --meta flag and default options (read length for this sample are on-average 51 bp, so I kept the default k-mer length used in metaSPAdes), and bowtie2 with --sensitive-local option.
*Thread Reply:* This might be an issue of using bowtie2 with --sensitive-local. The -local option allows for soft-clipping of reads and therefore, might clip off read ends that have damage because it is alignment-wise better than have three consecutive bases that have damage. You could try to use the global variant --sensitive and my guess you would have a proper damage signal.
*Thread Reply:* Thank you so much! It worked. So, if I understood correctly, for ancient metagenomic data assembly it is better to use the --sensitive flag instead of the --sensitive-local flag? It is a doubt that I had since I started this analysis, because I read some bibliography and it seemed that the --sensitive-local was widely used. When I used metaQuast to compare the completeness of the genomes (--sensitive-local vs --sensitive), the pipeline in which I used the --sensitive-local flag seemed to perform better.
*Thread Reply:* "--sensitive-local" is commonly used in most assembly pipelines for modern data. It usually doesn't matter for a lot of analyses, e.g. metaQUAST, but it has a particular effect when measuring the ancient DNA damage. @Maxime Borry had observed this before when he used nf-core/mag with default settings. It uses "--sensitive-local" and then the damage signal was gone.
*Thread Reply:* My 2 cents from Nepal: @Alex Hübner said everything, most likely soft clipping 🙂
*Thread Reply:* I wonder if this could be a little blog post... A few people have tripped up on this...
*Thread Reply:* No wait, add it to the little book of smiley plots!
https://www.spaam-community.org/little-book-of-smiley-plots/
*Thread Reply:* @Patrícia Santos would you mind sharing the analyze output table for just that contig/bin you have in the plot? Then someone can rerender it for the book and we can use Alex's description :)
*Thread Reply:* Yes, sure! It will be really helpful for people that are doing de-novo genome assembly for the first time :)
*Thread Reply:* Perfect thank you!
*Thread Reply:* Hi @Alex Hübner , @Patrícia Santos, and @Maxime Borry!
I finally got to adding this to the little book of smiley plots!
The PR is here if you can check if it looks OK: https://github.com/SPAAM-community/little-book-of-smiley-plots/pull/25
The page rendering are attached if you don't want to look at HTML and R code
If you're OK just say here or please leave a comment on Github (@Alex Hübner feel free to entirely re-write the description for phrasing and/or accuracy 😬, @Maxime Borry is the pyDamage extremely-simplified representation OK?)
<a href="https://github.com/SPAAM-community/little-book-of-smiley-plots">SPAAM-community/little-book-of-smiley-plots</a>


*Thread Reply:* OK I've got OKs from Alex and Patricia, @Patrícia Santos I will merge now, and share the link to the pages on the website. If you want any changes/suggestions let me know 🙂
*Thread Reply:* Softclipping page: https://www.spaam-community.org/little-book-of-smiley-plots/softclipping.html
*Thread Reply:* Contributors page: https://www.spaam-community.org/little-book-of-smiley-plots/contributors.html
*Thread Reply:* Ok @James Fellows Yates, thank you 🙂
*Thread Reply:* Thank you for sharing the plot and data 😄
*Thread Reply:* Now to find a artist 😄

Hi all! Do you know if there's a way to quantitatively assess the performance of taxonomic assignment produced by different algorithms based on phylogenetic clustering? Thanks!!
*Thread Reply:* Can you describe in more detail as to what you mean?
*Thread Reply:* Do you just mean compare accuracy of taxonomic assignment?
*Thread Reply:* Like comparing the topology?
*Thread Reply:* Hi @James Fellows Yates - Sorry I was not clear >.< For example, here I colored the nodes based on phylum-level identification. Amplicons in the same cluster share genetic similarity and so should have the same color. I would like to demonstrate that classification in the 2nd image, by showing a more cohesive color scheme, is likely doing a better job in taxonomic assignment compared to the one in the 1st image. Here it is only a visual representation, and I wonder if there is a quantitative measurement to support my statement.


*Thread Reply:* I see... I'm not sure myself... Maybe someone else has an idea?
Hi! I'm going to upload data to ENA and was wondering what "checklist" spreadsheet people have used the most? My samples are human and I will upload fastq files in most cases, but for some of the samples only bam files with only the reads mapping to certain microorganisms/bacteria. Any experiences? EDIT: The question is for "Register samples", I see that for uploading reads it's more straightforward but I assume I should start with registering samples?
*Thread Reply:* It's a little tricky for aDNA because of all of our samples are by default 'metagenomes'. But ENA doesn't seem to have the specific metagenome checklists versus mags etc)
I would thus go with the MIxS host- and/or human- associated, if they are from skeletons. But worst comes to worst can use the ENA default checklist
*Thread Reply:* Thanks @James Fellows Yates!

Hi all! I have a bioconda//docker question that I couldn't quite figure out from the online forums. Basically, I am wondering if is this a problem with package versions I'm using, or that I need to get in touch with my cluster to reconfigure my usr/local/src memory allocation (??).
I am trying to pull a docker image (https://hub.docker.com/r/vanearranz/mares/tags) via apptainer within a newly constructed environment on my local HPC cluster: ```### miniconda2
set up channel priorities
conda config --add channels defaults conda config --add channels bioconda conda config --add channels conda-forge
create environment with apptainer to pull docker image
conda create -n MARES_test -c conda-forge apptainer
enter environment
conda activate MARES_test
trying to pull MARES docker image
apptainer pull
and I got this error (there is more, but I wanted to just put up the fatal error bit)
FATAL: While making image from oci registry: error fetching image to cache: while building SIF from layers: packer failed to pack: while unpacking tmpfs: error unpacking rootfs: unpack layer: unpack entry: usr/local/src/taxdump/names.dmp: unpack to regular file: short write: write /tmp/build-temp-1152360637/rootfs/usr/local/src/taxdump/names.dmp: no space left on device``` If anyone has any something similar or a good direction I could look I would really appreciate it. Cheers!
*Thread Reply:* You've run out of space on your /tmp/ , normally that means your hpc doesn't use the default Linux /tmp/ space
*Thread Reply:* You can see that with the very last line
*Thread Reply:* ah yeah, thanks. I was a bit confused, so I wasn't sure if it was hpc issue or a package version issue. Thanks James! I hope you're having a great time at womad :headbangingparrot:
*Thread Reply:* Heh yeah, one of those cases you really need to read the entire error log and sort through all the cruft
Actually in the 'Jewel' at Singapore airport at the moment (left this morning)
Likely a very stupid "no-stupid-questions": We are asked to deposit our sequencing data to enable manuscript review and to provide "reviewer tokens". We would ideally fully release the data upon publication. However, ENA doesn't allow partial access, afaik. Do you know of other databases that could accomplish it or have any recommendations for how to navigate the request?
*Thread Reply:* It's NCBI related databases only that has that stuff...
*Thread Reply:* But I think you could argue that a) the ENA doesn't support this but it is a well established and commkn database for upload, b) if it is explicitly requested by reviewer your happy to share via a FTP server or something
*Thread Reply:* But it doesn't make sense to upload it twice, and nor upload to the wrong side of the world (internet speed reasons, and also submitting to ENA is easier)
*Thread Reply:* Thanks, James! That's very helpful (and speedy 🚀)!!!
*Thread Reply:* Or say you will release the data publicly if it's minor revisions 😬
*Thread Reply:* So likely to be accepted
*Thread Reply:* Hahahaha, way to say "want the data? just publish us!"

*Thread Reply:* Hi @Katerina Guschanski I got the same reviewer request and simply gave them the link to ENA saying they don't do that: https://ena-docs.readthedocs.io/en/latest/faq/release.html#can-i-make-my-study-partially-available
*Thread Reply:* Instead I provided the submission confirmation files, and that was enough for them
*Thread Reply:* Brilliant! Thank you @irinavelsko!!! That's a huge help. And I love the collective experience of SPAAM 😍
Hiya, lab related question. Has anyone managed to find nice storage boxes that properly fit eppendorf tubes into them? We usually use white cardboard boxes (which are also a poor fit for the tubes), but as I am at the start of a project, I had this lovely idea that I will colour code my freezer shelf with pink boxes for pellets, green for extracts and orange for libraries. So I ordered the boxes in the photo (https://www.starlabgroup.com/p/PF-SL-186914/product%2Fstarstore-81-storagebox-pf-sl-186914.html/I2381-5041). But the eppendorfs tubes don't fit properly in these boxes either because of the caps! It's such a stupid thing but it's driving me mad!!! Pls help 🙃


*Thread Reply:* Hi, we are also using these, leaving the last row and column empty.
*Thread Reply:* Thanks for the tip @Helja Niinemäe I'll try some rearranging next time I'm in the lab 🙂
*Thread Reply:* I’ve also used those. They’re not too bad - you just have to angle the caps diagonally and leave some empties along the edges

Hi! I am trying to run aMeta and I am getting this error at the BuildMaltDB, that I am not sure how to fix (shortened the paths to make it more readable): ```Job 14: BuildMaltDB: BUILDING MALT DATABASE USING SPECIES DETECTED BY KRAKENUNIQ Reason: Missing output files: results/MALT_DB/maltDB.dat
Activating conda environment: .snakemake/conda/214b9a1bcfb604a1ea11b90cad2330f0_
Activating conda environment: .snakemake/conda/214b9a1bcfb604a1ea11b90cad2330f0_
.snakemake/scripts/tmp1yl44jjf.malt-build.py:17: SyntaxWarning: invalid escape sequence '\d'
regex = re.compile("version (?P
RuleException:
CalledProcessError in file workflow/rules/malt.smk, line 26:
Command 'source /sw/apps/conda/latest/rackhamstage/bin/activate '.snakemake/conda/214b9a1bcfb604a1ea11b90cad2330f0'; set -euo pipefail; python .snakemake/scripts/tmp1yl44jjf.malt-build.py' returned non-zero exit status 1.
File "workflow/rules/malt.smk", line 26, in _ruleBuildMaltDB
File "aMeta_env/lib/python3.10/concurrent/futures/thread.py", line 58, in run``
It seems to start from one of the aMeta scripts (.snakemake/scripts/tmp1yl44jjf.malt-build.py`). Has anyone got the same error? How would you deal with it?
Thank you!!
*Thread Reply:* Hi @Markella Moraitou, thanks for using aMeta and I am ready to assist you with possible issues, do not hesitate post your questions here or direct message me. For now, could you post the log file from logs/BUILDMALTDB/BUILDMALTDB.log?
*Thread Reply:* Hi Nikolay, thanks for the quick response! The log file doesn't exist and neither does the BUILDMALTDB directory
*Thread Reply:* Hey @Markella Moraitou! This error is due to a problem with the new python versions and some syntax in the malt-build.py script. @Nikolay Oskolkov There is already an issue on Github about this error. https://github.com/NBISweden/aMeta/issues/155 I didn’t have time to work on a fix, but trying an older python version should work
<a href="https://github.com/NBISweden/aMeta">NBISweden/aMeta</a>
*Thread Reply:* Thank you so much! Apologies, I didn't think of checking the issues in GitHub!
*Thread Reply:* No worries! We are available on this channel and we should definitely fix that as soon as possible. Sorry about that! 😅
*Thread Reply:* Thanks @Zoé Pochon! So looks like we need to fix python3.10, right?
*Thread Reply:* @Markella Moraitou please try Zoe's suggestion and let us know it worked out
*Thread Reply:* Well, I guess we would need to adapt the syntax of the script so that python 3.10 and later versions don’t complain
*Thread Reply:* Ok, git it, let me check the syntax
*Thread Reply:* Not sure if I am doing something wrong, but I tried this with Python 3.11.4 and 3.9.5 and it fails again! I think the error message is different this time:
Activating conda environment: .snakemake/conda/214b9a1bcfb604a1ea11b90cad2330f0_
Activating conda environment: .snakemake/conda/214b9a1bcfb604a1ea11b90cad2330f0_
Traceback (most recent call last):
File ".snakemake/scripts/tmpocxh1tb0.malt-build.py", line 37, in <module>
shell(
File "aMeta_env/lib/python3.10/site-packages/snakemake/shell.py", line 294, in __new__
raise sp.CalledProcessError(retcode, cmd)
subprocess.CalledProcessError: Command 'set -euo pipefail; grep -wFf results/KRAKENUNIQ_ABUNDANCE_MATRIX/unique_species_taxid_list.txt aMeta_data/MALT/seqid2taxid.map.orig > results/MALT_DB/seqid2taxid.project.map; cut -f1 results/MALT_DB/seqid2taxid.project.map > results/MALT_DB/seqids.project; grep -Ff results/MALT_DB/seqids.project aMeta_data/MALT/library.fna.gz | sed 's/>//g' > results/MALT_DB/project.headers; seqtk subseq aMeta_data/MALT/library.fna.gz results/MALT_DB/project.headers > results/MALT_DB/library.project.fna 2>> logs/BUILD_MALT_DB/BUILD_MALT_DB.log; unset DISPLAY; malt-build -i results/MALT_DB/library.project.fna -a2t aMeta_data/MALT/nucl_gb.accession2taxid -s DNA -t 3 -d results/MALT_DB/maltDB.dat 2>> logs/BUILD_MALT_DB/BUILD_MALT_DB.log' returned non-zero exit status 1.
*Thread Reply:* @Markella Moraitou please post the log-file. If there is none or its empty, could you delete the whole folder ”results/MALT_DB” and restart aMeta?
*Thread Reply:* Thanks for responding so fast Nikolay! The log is indeed empty. The results/MALT_DB is not there either because the pipeline automatically deleted it after the error
Removing output files of failed job Build_Malt_DB since they might be corrupted:
results/MALT_DB/seqid2taxid.project.map, results/MALT_DB/seqids.project, results/MALT_DB/project.headers
Shutting down, this might take some time.
*Thread Reply:* I have run and re-run the pipeline quite a few times because I ran into different errors before (or had the job time out). I wonder if there would be any benefit in deleting all results and starting from zero.
*Thread Reply:* No, please do not delete the results so far
*Thread Reply:* Do you have a system log file maybe for the MALT_DB rule? I generally get a log in the log folder but also a log for the job from the system
*Thread Reply:* There is this log file.snakemake/log/2024-03-25T172216.737883.snakemake.log
I have been running it interactively using SLURM so I do not have a log file from the SLURM job, but this seems to include what I have been seeing in the terminal.
*Thread Reply:* @Markella Moraitou is your krakenuniqabundancematrix.txt non-empty?
*Thread Reply:* @Nikolay Oskolkov No, it is not empty
results/KRAKENUNIQ_ABUNDANCE_MATRIX/krakenuniq_abundance_matrix.txt
x
Acidovorax sp. YS12 218
Acinetobacter baumannii 507
Acinetobacter bouvetii 284
Acinetobacter cumulans 253
Acinetobacter defluvii 771
Acinetobacter indicus 1283
Acinetobacter johnsonii 850
Acinetobacter piscicola 18846
Acinetobacter schindleri 46
By the way, I am running this on a single sample as a test! I assume this shouldn't matter, but just in case!
*Thread Reply:* I think this shows that it is still using python 3.10:
/crex/proj/sllstore2017021/nobackup/MARKELLA/mammal_om_evolution/software/aMeta_env/lib/python3.10/concurrent/futures/thread.py", line 58, in run
*Thread Reply:* @Markella Moraitou especially if you are still in the first trials with the pipeline, I would indeed begin from 0 again and remove the conda aMeta environment using the conda command conda env remove -n aMeta and remove the aMeta folder. But keep your samples.tsv and config.yaml files preciously as well as your data files.
Then clone a new aMeta directory and open the file aMeta/workflow/envs/environment.yaml and change the line about python with that line: - python>=3.7,<3.10 . Then do the first step of the readme where you create the aMeta environment using mamba and replace your samples.tsv and config.yaml files where they should be. Then your aMeta environment will be using the python version on which aMeta should run bugfree 🤞. Then you can run again the lines from the README to recreate the rules environments and update the conda taxonomy. If you need help with this or something goes wrong, we can try to zoom next week. I'm sorry about that version problem. I also see that you are working from uppmax so I can provide you with a slurm config file to use the slurm queue with the pipeline.
Otherwise, if you can afford to wait a bit, I'll try to find a fix as soon as I find time (trying to wrap up a manuscript right now).
*Thread Reply:* Thanks @Zoé Pochon, I agree, a correct python version should be specified from the very beginning, i.e. when installing aMeta, so @Markella Moraitou I would try what Zoe suggests but would still copy the results-folder to a safe place just in case
*Thread Reply:* Thanks! I will try that! 🙂
*Thread Reply:* @Markella Moraitou I am working on the fix for the problem with building Malt database which you reported. So far I can't reproduce the error but we suspect it has something to do with the changed python syntax in the new python versions which makes malt-build.py fail. Do you by any chance have the log-file logs/BUILDMALTDB/BUILDMALTDB.log (apologies if I already asked for it, can't remember)? Or perhaps you have already managed to solve this issue by using an older python version?
*Thread Reply:* @Markella Moraitou we have a fix for your issue. This will be implemented centrally in aMeta soon, but in the meantime if you simply replace this line
regex = re.compile("version (?P<major>\d+)\.(?P<minor>\d+)")
with this line
regex = re.compile(r'version (?P<major>\d+)\.(?P<minor>\d+)')
in the aMeta/workflow/scripts/malt-build.py, this should fix the issue. Could you please try it and let me know whether it worked? Apologies for the issue, it came with some syntax changes in the recent Python versions and we had to adjust aMeta codes accordingly
*Thread Reply:* Hi Nikolay! Thank you so much, I will test this as soon as I can! :D Apologies for the silence after your last suggestion, I tried it but then I started running into other (possibly unrelated) issues so I left it aside for a bit! Unfortunately it seems like I deleted the previous output including the log files, but I will send the new one, if I get to the database building step without problems!
*Thread Reply:* Thanks @Markella Moraitou! We will certainly assist you with other issues as well, let us know here or DM me or Zoe
Hello everyone! Our lab has obtained several dental calculus samples, but we have no experience handling calculus. Since there are methods available that can recover both DNA and proteins, I would like to ask if you would extract both or only DNA?
*Thread Reply:* That's hard to say, depends on your question/what you want to do!
*Thread Reply:* I think the general takeaway is by doing both you will in one way or another get a lower DNA yield, which might not be great if you have bad preservation and your focus is on DNA
@Zandra Fagernäs @irinavelsko can correct me if I'm wrong as authors of one such protocol...
But calculs is generally well preserved so might not be an issue if your question you want to ask wants protein
*Thread Reply:* Thanks for your advice, James! If proteins are generally well preserved, then we need to pay more attention to the importance of protein extraction, even though we don't currently have the capacity for protein analysis. We will keep this in mind as we move forward with our research.🙏
*Thread Reply:* James is indeed correct! With well-preserved calculus, you will not see much loss in yield through a dual extraction protocol, but for poorly preserved samples you might. It's still better than splitting a sample in two and doing separate extractions though. But, as James said, the first question is what your research aim is, as the two biomolecules have slightly different strengths! 😊
*Thread Reply:* Thanks, Zandra! This is really helpful! Having both of your perspectives clarifies the trade-offs between protein and DNA yield depending on preservation quality and research focus. We'll definitely consider this when finalizing our protocol.

Hi! I was wondering if anyone has any recommendations for parasite specific databases. I've been using aMeta for my sample analysis but I'd like to look a bit more into parasite genomes and I don't really know where I should start
*Thread Reply:* Hi @Alicia Muriel, together with aMeta we provide a database called Microbial NT which includes 1.4 mln parasitic reference sequences:
Microbial NCBI NT database included 11,840,243 reference sequences (2,465,945 viral, 17,519 archaeal, 1,737,968 bacterial, 4,530,716 fungal, 1,689,877 protozoa, and 1,398,218 parasitic worms sequences)
*Thread Reply:* Thank you! I was wondering if I should use a more specific database but I’ll stick to that one then
*Thread Reply:* It may worth exploring whether you can use NCBI Datasets command line tools. You may be able to download parasites genomes but I’m not sure as I only used it to download arcahaea and bacterial genomes.
Hello everyone, I'm an M.Sc. student currently in an internship, and I'm working on ancient metagenomics data from dental calculus. I wanted to try aMeta for its resources' efficiency, but I am not familiar with SnakeMake and I struggle with the basic first steps. Could I ask you some questions ?
I'm trying to run the test provided with the workflow before running it on my data. I'm working on an HPC cluster that operates with SLURM. So I've filled the config/envmodules.yaml file like so :
envmodules:
fastqc:
- bioinfo/FastQC/0.12.1
cutadapt:
- bioinfo/Cutadapt/4.3
multiqc:
- bioinfo/MultiQC/1.19
bowtie2:
- bioinfo/bowtie/2.5.1
[...]
I didn't touch the config/config.yaml, and I used a profile for SLURM submissions wich is called smk-simple-slurm and added the --use-envmodules option . It looks like this :
cluster:
mkdir -p logs/{rule} &&
sbatch
--partition={resources.partition}
--qos={resources.qos}
--cpus-per-task={threads}
--mem={resources.mem_mb}
--job-name=smk-{rule}-{wildcards}
--output=logs/{rule}/{rule}-{wildcards}-%j.out
default-resources:
- partition=workq
- qos=inraeregion
- mem_mb=1000
restart-times: 3
max-jobs-per-second: 10
max-status-checks-per-second: 1
local-cores: 1
latency-wait: 60
jobs: 500
keep-going: True
rerun-incomplete: True
printshellcmds: True
scheduler: greedy
use-envmodules: True
So I'm firstly unsure if I have configured the workflow correctly.
I still tried to run the workflow from the .test/ directory with snakemake-v7.20.0 like so : snakemake --snakefile ../workflow/Snakefile -j 1 --profile simple-slurm/
I think it loads the module properly, but I have this error with the first FastQC step : Memory value 10240 MB was outside the allowed range (100 - 10000) at /usr/local/bioinfo/src/FastQC/FastQC-v0.12.1/fastqc line 203.
Should I adjust the memory allocated to FastQC, or is there a way to modify the allowed range?
Thank you in advance for taking the time to assist me. I am relatively new to workflow in general, especially SnakeMake ones, so I hope my questions aren't too trivial !
Best regards
*Thread Reply:* Hi @Mathias Caire, me and @Zoé Pochon will help you with aMeta. A quick fix for the FastQC "outside the allowed range" error is to change the default mem_mb in the aMeta/workflow/rules/qc.smk from its default 10240 to e.g. 1024 in the rules FastQC_BeforeTrimming and FastQC_AfterTrimming. Apologies for this silly bug, we will fix it centrally in aMeta asap.
*Thread Reply:* Regarding the configuration, I need to take a closer look, and I am at a meeting right now, so I will get back to you a bit later. In the meantime perhaps @Zoé Pochon could comment on your configuration?
*Thread Reply:* Hey @Mathias Caire! First, good job on putting that together. If it is running and it fails at the fastqc step it means that at least your files seems to work up to that step and there is no major error in them. Indeed, the fastqc rule has been changed recently in aMeta and since then it complains about the default range. You can change that in your smk-simple-slurm file like this:
```# Amount of threads per rule set-threads:
- FastQC_AfterTrimming=2
- FastQC_BeforeTrimming=2
Amount of resources per rule
set-resources:
- FastQCAfterTrimming:memmb=6000
- FastQCBeforeTrimming:memmb=6000``` Note that those are not tabs but spaces everywhere
*Thread Reply:* Thank you very much, the fastQC step now works ! I hope I will get the all thing to run soon with my data
*Thread Reply:* Hello ! Me again, I encounter an error with the filterign of krakenuniq outputs. Here is the message : ```Error in rule FilterKrakenUniqOutput: jobid: 15 input: results/KRAKENUNIQ/bar/krakenuniq.output, resources/pathogenomesFound.tab output: results/KRAKENUNIQ/bar/krakenuniq.output.filtered, results/KRAKENUNIQ/bar/krakenuniq.output.pathogens, results/KRAKENUNIQ/bar/taxID.pathogens, results/KRAKENUNIQ/bar/taxID.species log: logs/FILTERKRAKENUNIQOUTPUT/bar.log (check log file(s) for error details) conda-env: /work/project/GenIn/ROUSSILLE/aMeta/.test/.snakemake/conda/6e63d658e3ebb26afeed2f78cbd5f25d_ shell: /work/project/GenIn/ROUSSILLE/aMeta/workflow/scripts/filterkrakenuniq.py results/KRAKENUNIQ/bar/krakenuniq.output 1000 200 resources/pathogenomesFound.tab &> logs/FILTERKRAKENUNIQOUTPUT/bar.log; cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.pathogens | tail -n +2 > results/KRAKENUNIQ/bar/taxID.pathogens;cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.filtered | tail -n +2 > results/KRAKENUNIQ/bar/taxID.species (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!) clusterjobid: Submitted batch job 7020974
Error executing rule FilterKrakenUniqOutput on cluster (jobid: 15, external: Submitted batch job 7020974, jobscript: /work/project/GenIn/ROUSSILLE/aMeta/.test/.snakemake/tmp.ff2hgy7s/snakejob.FilterKrakenUniqOutput.15.sh). For error details see the cluster log and the log files of the involved rule(s).``
The weird thing is that there is no log file in theFILTERKRAKENUNIQOUTPUT` folder so I can't identify where the error comes from... I was thinking that you may have an idea about this issue. Thank you in advance for taking the time to assist me.
*Thread Reply:* Hey Mathias!
Let's rule out a time-limit or memory issue first.
Can you write on your terminal
sacct -j 7020974
and
seff 7020974
*Thread Reply:* ```JobID JobName Partition Account AllocCPUS State ExitCode
7020974 smk-Filte+ workq mcaire 1 FAILED 1:0 7020974.bat+ batch mcaire 1 FAILED 1:0 7020974.ext+ extern mcaire 1 COMPLETED 0:0```
*Thread Reply:* Okay, then something went wrong. What do you have in your aMeta/results/KrakenUniq/**/**folder?
*Thread Reply:* I just threw the output in the chat sorry, I sent it by accident haha In my KrakenUniq output I think I have the right files, here they are : ```==> bar/krakenuniq.output <==
KrakenUniq v1.0.4 DATE:20240422T13:44:43Z DB:resources/KrakenUniqDB DBSIZE:99800462 WD:/work/project/GenIn/ROUSSILLE/aMeta/.test
CL:/usr/local/bioinfo/src/KrakenUniq/krakenuniq-v1.0.4/krakenuniq --preload --db resources/KrakenUniqDB --fastq-input results/CUTADAPTADAPTER_TRIMMING/bar.trimmed.fastq.gz --threads 10 --output results/KRAKENUNIQ/bar/sequences.krakenuniq --report-file results/KRAKENUNIQ/bar/krakenuniq.output --gzip-compressed --only-classified-out
% reads taxReads kmers dup cov taxID rank taxName 55.64 710 710 33190 1.04 NA 0 no rank unclassified 44.36 566 0 17686 1 0.001772 1 no rank root 44.36 566 0 17686 1 0.001772 131567 no rank 40.13 512 0 15898 1 0.00177 2 superkingdom Bacteria 40.13 512 0 15898 1 0.00177 1224 phylum 40.13 512 0 15898 1 0.00177 1236 class 40.13 512 0 15898 1 0.00177 91347 order
==> bar/sequences.krakenuniq <== C endo542:+:3651:3702:51e10-2/1 632 51 632:16 0:15 C endo830:-:5112:5155:43e10-2/1 632 43 0:14 632:9 C endo113:+:3286:3332:46e10-2/1 632 46 632:1 0:21 632:4 C endo410:+:4001:4061:60e10-2/1 632 60 0:15 632:25 C endo492:+:697:738:41e10-2/1 632 41 0:8 632:13 C endo130:-:805:849:44e10-2/1 632 44 632:5 0:19 C endo156:+:2707:2745:38e10-2/1 632 38 632:18 C endo916:-:1531:1587:56e10-2/1 9605 56 9605:17 0:19 C endo863:+:7062:7132:70e10-2/1 632 70 632:15 0:21 632:14 C endo445:+:8065:8147:82e10-2/1 632 75 632:16 0:21 632:18
==> foo/krakenuniq.output <==
KrakenUniq v1.0.4 DATE:20240422T13:43:57Z DB:resources/KrakenUniqDB DBSIZE:99800462 WD:/work/project/GenIn/ROUSSILLE/aMeta/.test
CL:/usr/local/bioinfo/src/KrakenUniq/krakenuniq-v1.0.4/krakenuniq --preload --db resources/KrakenUniqDB --fastq-input results/CUTADAPTADAPTER_TRIMMING/foo.trimmed.fastq.gz --threads 10 --output results/KRAKENUNIQ/foo/sequences.krakenuniq --report-file results/KRAKENUNIQ/foo/krakenuniq.output --gzip-compressed --only-classified-out
% reads taxReads kmers dup cov taxID rank taxName 44.57 608 608 31393 1.05 NA 0 no rank unclassified 55.43 756 0 23399 1 0.002345 1 no rank root 55.43 756 0 23399 1 0.002345 131567 no rank 49.63 677 0 20950 1 0.002332 2 superkingdom Bacteria 49.63 677 0 20950 1 0.002332 1224 phylum 49.63 677 0 20950 1 0.002332 1236 class 49.63 677 0 20950 1 0.002332 91347 order
==> foo/sequences.krakenuniq <== C endo735:-:9954:9993:39e10-2/1 632 39 632:5 0:14 C endo752:+:2727:2828:101e10-2/1 632 75 632:55 C endo516:+:8375:8423:48e10-2/1 632 48 632:28 C endo46:-:4794:4997:203e10-2/1 632 75 632:45 0:10 C endo54:+:7210:7299:89e10-2/1 632 75 632:14 0:21 632:20 C endo795:+:1290:1415:125e10-2/1 632 75 632:55 C endo417:+:867:920:53e10-2/1 632 53 0:16 632:17 C endo72:-:7405:7476:71e10-2/1 632 71 632:51 C endo88:-:3241:3280:39e10-2/1 632 39 632:19 C endo986:-:5062:5107:45e10-2/1 9605 45 9605:23 0:2 ```
*Thread Reply:* And you said that there is no log file there? logs/FILTERKRAKENUNIQOUTPUT/bar.log
*Thread Reply:* Yes FILTERKRAKENUNIQOUTPUT is an empty directory, that's strange no ?
*Thread Reply:* Hmm. There should be at least a log from slurm. I see that in your config file, you tell slurm to save the logs this way: --output=logs/{rule}/{rule}-{wildcards}-%j.out Can you try to look if that one is there?
*Thread Reply:* Should be logs/FilterKrakenUniqOutput/FilterKrakenUniqOutput**
*Thread Reply:* I have some files but they simply contain the standard output of the workflow : ```[Mon Apr 22 15:51:50 2024] Job 0: FilterKrakenUniqOutput: APPLYING DEPTH AND BREADTH OF COVERAGE FILTERS TO KRAKENUNIQ OUTPUT FOR SAMPLE results/KRAKENUNIQ/bar/krakenuniq.output resources/pathogenomesFound.tab Reason: Missing output files: results/KRAKENUNIQ/bar/krakenuniq.output.pathogens, results/KRAKENUNIQ/bar/taxID.pathogens, results/KRAKENUNIQ/bar/krakenuniq.output.filtered, benchmarks/FILTERKRAKENUNIQOUTPUT/bar.benchmark.txt, results/KRAKENUNIQ/bar/taxID.species
/work/project/GenIn/ROUSSILLE/aMeta/workflow/scripts/filterkrakenuniq.py results/KRAKENUNIQ/bar/krakenuniq.output 1000 200 resources/pathogenomesFound.tab &> logs/FILTERKRAKENUNIQOUTPUT/bar.log; cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.pathogens | tail -n +2 > results/KRAKENUNIQ/bar/taxID.pathogens;cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.filtered | tail -n +2 > results/KRAKENUNIQ/bar/taxID.species Activating environment modules: bioinfo/KrakenUniq/1.0.4 /tools/tools/modules/4.6.1/init/bash: line 88: deactivate: command not found [Mon Apr 22 15:51:51 2024] Error in rule FilterKrakenUniqOutput: jobid: 0 input: results/KRAKENUNIQ/bar/krakenuniq.output, resources/pathogenomesFound.tab output: results/KRAKENUNIQ/bar/krakenuniq.output.filtered, results/KRAKENUNIQ/bar/krakenuniq.output.pathogens, results/KRAKENUNIQ/bar/taxID.pathogens, results/KRAKENUNIQ/bar/taxID.species log: logs/FILTERKRAKENUNIQOUTPUT/bar.log (check log file(s) for error details) conda-env: /work/project/GenIn/ROUSSILLE/aMeta/.test/.snakemake/conda/6e63d658e3ebb26afeed2f78cbd5f25d shell: /work/project/GenIn/ROUSSILLE/aMeta/workflow/scripts/filterkrakenuniq.py results/KRAKENUNIQ/bar/krakenuniq.output 1000 200 resources/pathogenomesFound.tab &> logs/FILTERKRAKENUNIQ_OUTPUT/bar.log; cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.pathogens | tail -n +2 > results/KRAKENUNIQ/bar/taxID.pathogens;cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.filtered | tail -n +2 > results/KRAKENUNIQ/bar/taxID.species (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Shutting down, this might take some time. Exiting because a job execution failed. Look above for error message```
*Thread Reply:* Hmm, it is tricky to find the error without the log 🧐
*Thread Reply:* Is there maybe a problem with one of your modules?
/tools/tools/modules/4.6.1/init/bash: line 88: deactivate: command not found
*Thread Reply:* I don't normally get that warning/error
*Thread Reply:* I don't know, since I use the --use-env-modules option I thought it was normal, what do you think ?
*Thread Reply:* I just found another strange thing, when I simply try to run :
/work/project/GenIn/ROUSSILLE/aMeta/workflow/scripts/filter_krakenuniq.py results/KRAKENUNIQ/bar/krakenuniq.output 1000 200 resources/pathogenomesFound.tab &> logs/FILTER_KRAKENUNIQ_OUTPUT/bar.log; cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.pathogens | tail -n +2 > results/KRAKENUNIQ/bar/taxID.pathogens;cut -f7 results/KRAKENUNIQ/bar/krakenuniq.output.filtered | tail -n +2 > results/KRAKENUNIQ/bar/taxID.species
There is no error and I have the output files like this :
```==> results/KRAKENUNIQ/bar/krakenuniq.output <==
KrakenUniq v1.0.4 DATE:20240422T13:44:43Z DB:resources/KrakenUniqDB DBSIZE:99800462 WD:/work/project/GenIn/ROUSSILLE/aMeta/.test
CL:/usr/local/bioinfo/src/KrakenUniq/krakenuniq-v1.0.4/krakenuniq --preload --db resources/KrakenUniqDB --fastq-input results/CUTADAPTADAPTER_TRIMMING/bar.trimmed.fastq.gz --threads 10 --output results/KRAKENUNIQ/bar/sequences.krakenuniq --report-file results/KRAKENUNIQ/bar/krakenuniq.output --gzip-compressed --only-classified-out
% reads taxReads kmers dup cov taxID rank taxName 55.64 710 710 33190 1.04 NA 0 no rank unclassified 44.36 566 0 17686 1 0.001772 1 no rank root 44.36 566 0 17686 1 0.001772 131567 no rank 40.13 512 0 15898 1 0.00177 2 superkingdom Bacteria 40.13 512 0 15898 1 0.00177 1224 phylum 40.13 512 0 15898 1 0.00177 1236 class 40.13 512 0 15898 1 0.00177 91347 order
==> results/KRAKENUNIQ/bar/krakenuniq.output.filtered <== % reads taxReads kmers dup cov taxID rank taxName 40.13 512 512 15898 1.0 0.00177 632 species Yersinia pestis
==> results/KRAKENUNIQ/bar/krakenuniq.output.pathogens <== % reads taxReads kmers dup cov taxID rank taxName 40.13 512 512 15898 1.0 0.00177 632 species Yersinia pestis
==> results/KRAKENUNIQ/bar/sequences.krakenuniq <== C endo542:+:3651:3702:51e10-2/1 632 51 632:16 0:15 C endo830:-:5112:5155:43e10-2/1 632 43 0:14 632:9 C endo113:+:3286:3332:46e10-2/1 632 46 632:1 0:21 632:4 C endo410:+:4001:4061:60e10-2/1 632 60 0:15 632:25 C endo492:+:697:738:41e10-2/1 632 41 0:8 632:13 C endo130:-:805:849:44e10-2/1 632 44 632:5 0:19 C endo156:+:2707:2745:38e10-2/1 632 38 632:18 C endo916:-:1531:1587:56e10-2/1 9605 56 9605:17 0:19 C endo863:+:7062:7132:70e10-2/1 632 70 632:15 0:21 632:14 C endo445:+:8065:8147:82e10-2/1 632 75 632:16 0:21 632:18
==> results/KRAKENUNIQ/bar/taxID.pathogens <== 632
==> results/KRAKENUNIQ/bar/taxID.species <== 632```
*Thread Reply:* Cool! I'm honestly not sure what is causing that problem. Sometimes snakemake understands as a "fail" when there is a warning message produced. Did you get any warning when running it manually?
And do you get that warning:
/tools/tools/modules/4.6.1/init/bash: line 88: deactivate: command not found
In other slurm log files of completed jobs?
*Thread Reply:* I didn't get any warnings but I'm glad that it's working, now if I rerun the workflow the following steps should start right ?
I do get the deactivate: command not found everywhere, even the completed jobs
*Thread Reply:* Yes I think it will start from there. You can do a dry run by adding -n to your snakemake command before running it for real and you will see which jobs it is planning on running
*Thread Reply:* Thank you for the help !
*Thread Reply:* No worries! Fingers crossed 😊🤞
*Thread Reply:* Hello !
I come back to ask questions about aMeta because I'm I'm struggling with the resources that I need. The workflow works really well with my data when I use the microbial part of NCBI RefSeq database that you provided on scilifelab. However, I would like to use a more comprehensive one.
I downloaded the Refeq archaea, bacteria, viral, plasmid, human1, UniVec_Core, protozoa & fungi from the Kraken 2 indexes repository here https://benlangmead.github.io/aws-indexes/k2. But the index is in this format :
3.9M database100mers.kmer_distrib
3.6M database150mers.kmer_distrib
3.3M database200mers.kmer_distrib
3.0M database250mers.kmer_distrib
2.8M database300mers.kmer_distrib
4.5M database50mers.kmer_distrib
4.2M database75mers.kmer_distrib
77G hash.k2d
3.2M inspect.txt
59G k2_pluspf_20240112.tar.gz
2.4M ktaxonomy.tsv
44M library_report.tsv
512 opts.k2d
3.7M taxo.k2d
8.0K unmapped_accessions.txt
Which doesn't work when I just use the folder containing it in aMeta.
Do I need to take additional steps for KrakenUniq to utilize it?
Additionally, I utilized the option to screen for pathological taxa with Bowtie2 but I cannot find where the results are after running the workflow.
Thank you in advance for your help !
Best regards Mathias CAIRE
*Thread Reply:* Hi @Mathias Caire, aMeta is very KrakenUniq-centric and does not work with Kraken2 database. We did not want to use Kraken2 for two reasons. First, higher RAM demands of Kraken2 compared to KrakenUniq. Second, the breadth of coverage filter (number of unique kmers) was initially developed in KrakenUniq and imported later to Kraken2, so at the moment of releasing first versions of aMeta, Kraken2 did not have that filter. In summary you will need a Kraken1 or KrakenUniq database to make aMeta work. I would recommend you to download the Microbial NT database which is actually more comprehensive than the standard Kraken2 database, this was shown in the aMeta paper
*Thread Reply:* Thank you for the response, I will follow your advice and run aMeta on the Microbial NT Database. Could you also assist me with pathogen screening using Bowtie2? What output is expected for this step of the workflow?
*Thread Reply:* Oh, sorry @Mathias Caire, forgot to answer. In the results/KRAKENUNIQ folder for each sample you have a “.pathogens” files which represent an overlap of krakenuniq.filtered with a custom very permissive list of known pathogens. So you can check which of your detected microbes can be potential pathogens. In earlier versions of aMeta, only reads corresponding to those pathogenic microbes were aligned to their references with Bowtie2, and their ancient status was checked with mapDamage. Now this is done for all detected microbes, including the pathogenic microbes. The Bowtie2 branch of aMeta served as a quick and dirty way to get alignments+deamination. Ideally one should do it via Malt which has an LCA, i.e. a more clever and metagenomics-specific way of alignment. However, since Malt can be a headache, while waiting for Malt results, one might want to get quick preliminary results from the Bowtie2 analysis
*Thread Reply:* Thank you very much! I do indeed have the file you mentioned. I must say that I really like the workflow, and I'm impressed with how the resources are handled. Have a nice day!
*Thread Reply:* Great to hear @Mathias Caire, thank you!
*Thread Reply:* Hello, I have some more questions if you have the time to answer them. I need to run some analysis on the results, but for that, I require the OTU table containing the raw abundance of each taxa in my samples. I believe I should use the MALT abundance matrix, but I'm unclear about the differences between the SAM and RMA6 versions.
Before transitioning to aMeta, I had already conducted MALT analysis on my samples and I was processing the RMA6 files with MEGAN. I could compare them using MEGAN options and export an OTU table along with a tree in Newick format (both extremely helpful for my subsequent analysis). I've attempted to replicate this process with the outputs in the MALT directory, but I'm struggling to obtain the same matrix as the maltabundancematrix_rma6.
What are your thoughts on this?
Best regards, Mathias CAIRE
*Thread Reply:* Hi @Mathias Caire, you most likely need to use the RMA6 abundance matrix which should be equivalent to the MEGAN abundance matrix which you used previously. The SAM abundance matrix was provided mainly for an "upper limit estimate" of the microbial abundance (i.e. likely an overestimate) as a possible way to quantify SAM-alignments provided by MALT.
Since MALT outputs two types of alignments: RMA6 (bioinformatically unfriendly) and SAM (bioinformatically friendly), we wanted to look inside both of them. The difference between the RMA6 and SAM abundance matrices is that the former is computed from RMA6 alignments (with LCA, more appropriate for metagenomic samples), and the latter is computed from SAM-alignments (without LCA, aka regular Bowtie2 alignments). The lack of LCA will most likely result in accounting for multi-mappers and therefore an overestimate of the true abundance, while the RMA6 (with LCA) abundance may be too conservative, i.e. ignoring too many valuable reads (especially for low-coverage samples) which it treats as multi-mappers. Still I would perhaps recommend to stick to the RMA6 abundance and use the SAM as a way to double-check the microbes which you found interesting from the RMA6 table
*Thread Reply:* Thank you for answering so quickly, that helps a lot. I also need a tree in Newick format but I think I can manage to extract it from the ncbi.tre file and the taxon list.
*Thread Reply:* Hmm, to get a Newick tree you should probably input the RMA6-alignments from a Malt job to MEGAN, i.e. outside of aMeta
*Thread Reply:* I cannot mannage to get the same species as in the RMA6 abundance matrix. For now I simply loaded all the RMA6 in MEGAN and used the optioin "compare" of MEGAN and selected all of them. I uncollapsed the tree and I selected the species to extract the tree. Should I do something differently ?
*Thread Reply:* Hmm @Mathias Caire, for quantifying microbial abundance on species level from rma6-files we use rma-tabuliser tool from @James Fellows Yates. The species count table from rma-tabuliser should be more or less equivalent to the rma6-files which you can visualize in MEGAN, i.e. the species names at least should be the same, right James?
*Thread Reply:* I may have caused some confusion. I initially thought that the species represented in the heatmap were the same as those in the RMA6 matrix. When I mentioned that I couldn't find the same species, I meant that some species present in the heatmap are missing from the table I generated using MEGAN, and vice versa.
I think I got a little bit confused because the RMA6 matrix looks like this :
node SP1 SP2 SP3
1 ... ... ...
2 ... ... ...
29 ... ... ...
31 ... ... ...
Are those nodes the taxIDs? If so, how can I obtain their taxonomical names? Currently, I'm running a script that searches through a taxDB file to find the taxa names, but I am finding various taxonomical ranks (genus, order, etc.) rather than just species as i would expect in the RMA6 matrix.
*Thread Reply:* @Mathias Caire the species in the heatmap are usually a subset of those in RMA6. This is because not all detected species (i.e. the ones in the RMA6 file) can be successfully authenticated. So the heatmap represents the most robust species which passed multiple validation and authentication steps and got the authentication score (from 0 to 10), which is displayed in the heatmap.
Yes, the first column in the maltabundancematrixrma6.txt are taxIDs. You can get their corresponding scientific names scientific names if you navigate to aMeta/results/KRAKENUNIQABUNDANCE_MATRIX and type
paste unique_species_taxid_list.txt unique_species_names_list.txt
*Thread Reply:* Btw thank you for reminding, it has been a long-standing plan to make it more clear for the users. I will fix it now asap in the next release
*Thread Reply:* @Nikolay Oskolkov thank you very much for your help, I'm glad I can contribute to make aMeta more user-friendly. Have a great day!
Hi y'all,
a while ago somebody held a super interesting talk about detection of false positivehits in (in metagenomic screenings), I believe to remember that it was on the example of parasites and/or pathogens. It was also reassiesing some older published material. I would be very glad if someone remembers more closely and can give me the the link to the paper.
If someone has generally more literature on identification methods for false positive hits to share, I would be interested as this topic seems to pop up more and more recently.
All the best :)
*Thread Reply:* Hi @Freya Steinhagen, I have have my private collection of false-positive hits in metagenomic analyses, and if you want we could talk about it in more details (e.g. via zoom). I do not think there are many publications on this topic, but I could immediately recommend this very good review https://www.annualreviews.org/content/journals/10.1146/annurev-genom-091416-035526 and especially the fantastic supplementary figure 1 from that paper https://www.annualreviews.org/docserver/fulltext/genom/18/1/gg18warinnersupfigures.pdf?expires=1713350923&id=id&accname=ar-269936&checksum=6CEFB66D33511B2F24A760D418E20F29|https://www.annualreviews.org/docserver/fulltext/genom/18/1/gg18warinnersupfigures.pd[…]ccname=ar-269936&checksum=6CEFB66D33511B2F24A760D418E20F29, which shows how easy it is to "discover" a pathogen in a soil or ocean if you are doing a naive analysis. Also, a large chunk of our aMeta paper https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03083-9 (see Figure 9) was about false-positive discoveries and how aMeta attempts to combat them

*Thread Reply:* Also, you might want to check this https://dgg32.medium.com/carp-in-the-soil-1168818d2191
*Thread Reply:* Besides Nicholay 's, is the speech "Parasit detection in ancient and modern metagenomic datasets" from Jonas Niemann?
*Thread Reply:* Yes, thanks! that might be the one I was thinking of
*Thread Reply:* @Wenqin Yu and @Freya Steinhagen do you have the reference for the "Parasite detection..."?
*Thread Reply:* Also a good read from @Aleksandra Laura Pach https://www.biorxiv.org/content/10.1101/2024.02.27.581519v1
*Thread Reply:* @Jonas Niemann hopefully something soon?
*Thread Reply:* @Nikolay Oskolkov Sorry I can't find it as well, it seems an unpublished work.
*Thread Reply:* Thanks, I also realized that it was (apparently) a SPAAMtisch talk that I probably missed. @Maria Lopopolo are all the SPAAMtisch talks recorded and available? Could you post a link? It was my long-term plan to watch some talks that I missed
*Thread Reply:* Hello, yes all the talks from speakers who gave me consent are recorded. Most of them are on the SPAAM you tube channel. Unfortunately the parasite one was one of the few I could not record for the reason that I did not receive consent from the speaker. But I can ask the speaker if we can open a Google doc with questions you may have for them. Do you think it can help?
*Thread Reply:* Thanks Maria for the great job you and others are doing within the SPAAMtisch initiative! I could see that very few SPAAMtisch talks were present at the SPAAM youtube channel and thought there was another channel. But if the speaker did not want the talk to be recorded, it is fine, no need to bother 🙂

Hi all, I have gotten 0.1 and 0.05 as 2 damage frequencies at the read end for different species from the same ancient metagenomic sample. The screenshots are 2 species for example. There has been a discussion for whether the one with lower damage frequency (0.05) should be considered as authenticated or with a clear damage pattern. Do you think the lower one is a result of the probability of DNA damage and stochasticity of reads, or it shall not be considered authenticated?


*Thread Reply:* The extent of a recovered aDNA damage signal is impacted by divergence from the reference sequence. In this case, the species on the left is more diverged (non-damage substitution rates are higher), which might be limiting the damage signal.
*Thread Reply:* Depends on the level of degradation too (e.g. age,other preservation factors). To me the lower damage one would still be valid, the curve is relatively smooth rather than spiky and inconsistent
You could try running it against @Maxime Borry's PyDamage if you want a statistical check
*Thread Reply:* Against a model
*Thread Reply:* If you look at @Katerina Guschanski and @Jaelle Brealey's 200y old bear calculus microbial genomes, iirc they look quite similar to your lower ok nes
*Thread Reply:* If you were to fix the Y axis it would also help evaluate the difference from reference: you would expect to see the base line of all other substitutions to be quite high if you have the wrong or diverted reference (right @Pete Heintzman?)
*Thread Reply:* Thank you Pete and James.
I ran Pydamage with default setting and it passed all the species (even the spiky ones). I am looking for the original script right now.
I will read the paper of Katja and Jaelle ❤️
*Thread Reply:* The level of damage would be consistent with younger (museum-age, a few hundred years) samples. The baseline, which as @Pete Heintzman suggests would be indicative to distance to the reference genomes, actually looks quite similar to me for both examples, so I'm not sure if it would explain the difference in damaged proportion. I would tend to agree that the left-hand example looks genuine
Where can I find the fasta sequences from the AMDirT? I want them (just the viral ones) for my DB and I can not find them in the NCBI neither on EMBI-EBI, I have the ids as DA66 for ERS2484289 DA337 ERS2484292... any advice?
*Thread Reply:* AMDirT only supports FASTQ files at the moment, as per the warning, as otherwise the way to download the data are too heterogeneous so its not easy to implement.
I guess though the FASTA sequences can be found via the ENA so the warning isn't very precise and we should improve that.
Also add that caveat to the documentation now I think about it (ping @Maxime Borry)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
*Thread Reply:* You'll have to inspect the tables manually to get to get the FASTAs... Sorry @Ania TSL :(
*Thread Reply:* @Ania TSL if you check the sample table ancientsinglegenome-hostassociated and filter for the sample name DA66, you see at the far end of the table that the archive is GitHub. The authors never uploaded their data to either NCBI or ENA but just kept them on GitHub.
I guess the sequences you search for are here: https://github.com/acorg/parvo-2018/blob/master/consensuses/consensuses.fasta
<a href="https://github.com/acorg/parvo-2018">acorg/parvo-2018</a>
*Thread Reply:* Thanks Alex!
*Thread Reply:* Thank you so much for your replies. Also @Miriam Bravo show me how to download them from the ENA with the project number and is working.
Hi I had a quick question that I hoped someone here could help me with, does anyone know where to find the SNP Evaluation documentation? I've checked the github repo but can't seem to find it. There is a powerpoint but it details a problem and a fix for the program but they don't seem to have any information on usage or how the program actually works.
only thing I could find was an active issue opened in 2019 saying the link to the pdf manual is broken:
https://github.com/andreasKroepelin/SNP_Evaluation/issues/1
If anyone knows where to find the documentation or could point me in the right direction it would be greatly appreciated. 🙂
<a href="https://github.com/andreasKroepelin/SNP_Evaluation">andreasKroepelin/SNP_Evaluation</a>
*Thread Reply:* Last time I asked, it didn't exist. But if I remember correctly from the last (and only) time I used it, there were help pages available in the help menu. Maybe @aidanva or @Alina Hiss knows more details
*Thread Reply:* If you go to the “?” in the menu, there is a bit of information on how to use it. But it is not a lot. I will suggest you get into contact with Alexander Herbig (alexander_herbig@eva.mpg.de), to see if he has a copy of this mysterious PDF.
*Thread Reply:* Thanks, its a shame that this is turning into a much more difficult task then I'd hoped. I was mostly interested in how the program decided what is and ins’t a false positive SNP.
A paper I was looking at used it and based off the output table in the supplementary material it wasn’t clear to me how and why SNPs had or hadn’t been classified as false positives by the program so I was curious where this score was coming from 😅.
*Thread Reply:* The score is based on the different metrics you can calculate, one of them being able to compare more strict mapping vs less strict mapping and seeing how the coverage is affected, you can also calculate the number of heterozygous calls around the SNP and also the number of bases missing in a a window around the SNP
*Thread Reply:* Thanks after going back to the table and carefully looking through I reached the same conclusion. I noticed that only SNPs with a score of 1 were deemed to be not due to contamination and the score equaled the sum of the mapping ratio (50bp window), heterozygous positions (50bp window) and positions not covered (50bp window).
Meaning only SNPs that didn’t have any heterozygous calls in a 50 bp window, had all positions in a 50bp window covered and the mean coverage of the lenient mapping and strict mapping in a 50bp window was identical were considered not to be due to contamination.
*Thread Reply:* The paper was Spyrou et al 2019 so not the most recent paper: https://www.nature.com/articles/s41467-019-12154-0
*Thread Reply:* yeah, exactly, that’s how you should interpret the score 🙂
*Thread Reply:* you might also want to check Keller et al. 2019: Ancient Yersinia pestis genomes from across Western Europe reveal early diversification during the First Pandemic www.pnas.org/cgi/doi/10.1073/pnas.1820447116 although i am not sure if it gives more info than what has been said here before

Hi All! Have a quick wet lab question: we’re looking at Quantas Quantiflor as an alternative to Qubit HS for quantification. Unsure if they’re comparable in performance for low concentration. Anyone used Quantas/found it as good as Qubit?
Hi there, I am trying to establish analytical pipelines on a new cluster. I am doing it from scratch for the first time and I am running into some issues. Currently, I am trying to install aMeta @Zoé Pochon and when I run the test I do not get the full set of results with plots etc. What is supposed to be the output from the test run?
*Thread Reply:* @Joanna H. Bonczarowska please check this section in the aMeta github to learn about the main output files https://github.com/NBISweden/aMeta?tab=readme-ov-file#main-results-of-the-workflow-and-their-interpretation
*Thread Reply:* I only have "CUTADAPTADAPTERTRIMMING" directory with fastq files foo and bar in the ./test/results So I guess it means that the test run failed and did not generate all the output?
*Thread Reply:* Yes, possibly. Snakemake usually throws a lot of red text when an error occurs. Also at the very final lines of snakemake output it gives a link to a overall log-file, could you please post it here?
*Thread Reply:* I submitted the runtest.sh as a batch job in a cluster and I did not get any information printed out into a log file anywhere
*Thread Reply:* A batch job should typically produce a log-file of everything it would write on a screen if it was executed locally.
*Thread Reply:* Perhaps for installation purposes I would recommend to book a node via an interactive job. This way (when you control all the messages) the installation will be more efficient
*Thread Reply:* Hey Joanna! Indeed, I think it might be best to run the test jobs directly on the login node or if this is strictly forbidden to book an interactive session of some cores to run the test and directly see the error messages. Maybe you could add the option - - keep-going in the Snakemake command within the runtest script so that it doesn't stop at the first small error
*Thread Reply:* I am now running it as you suggested, in an interactive session, and I got this error:
*Thread Reply:* Error in rule FastQCBeforeTrimming: jobid: 3 input: data/bar.fq.gz output: results/FASTQCBEFORETRIMMING/barfastqc.html, results/FASTQCBEFORETRIMMING/barfastqc.zip log: logs/FASTQCBEFORETRIMMING/bar.log (check log file(s) for error details) conda-env: /gpfs/gibbs.../.test/.snakemake/conda/c6ec736af08d79289f3602ae7da070b7 shell: fastqc data/bar.fq.gz --memory 10240 --threads 1 --nogroup --outdir results/FASTQCBEFORETRIMMING &> logs/FASTQCBEFORETRIMMING/bar.log (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Logfile logs/FASTQCBEFORETRIMMING/bar.log:
Memory value 10240 MB was outside the allowed range (100 - 10000) at /gpfs/gibbs/project/.../.test/.snakemake/conda/c6ec736af08d79289f3602ae7da070b7_/bin/fastqc line 203.
*Thread Reply:* I tried to both increase and decrease the memory for the job but it did not help
*Thread Reply:* Yes, a known bug. To be fixed very soon
*Thread Reply:* Is there a way to go around it?
*Thread Reply:* For the moment I only specify the amount of cores and memory in my slurm config file like this:
Amount of threads per rule
set-threads:
- FastQC_AfterTrimming=2
- FastQC_BeforeTrimming=2
Amount of resources per rule
set-resources:
- FastQCAfterTrimming:memmb=6000
- FastQCBeforeTrimming:memmb=6000
*Thread Reply:* Yes, please try Zoe’s suggestion, and also please check here https://github.com/NBISweden/aMeta/issues/156
<a href="https://github.com/NBISweden/aMeta">NBISweden/aMeta</a>
*Thread Reply:* But I don't really know how to implement that within the test run 🤔
*Thread Reply:* In the ./tes/config.yaml?
*Thread Reply:* Ok that did not work 💀
*Thread Reply:* Please open aMeta/workflow/rules/qc.smk and change one line in the rules FastQCAfterTrimming and FastQCBevoreTrimming
*Thread Reply:* please remove 0 at the end
*Thread Reply:* Yeah, what Nikolay is suggesting is probably the easiest
*Thread Reply:* there were two lines with mem_mb=10240, I changed them both (when I changed just the first one it did not work). Now it seems to be running!
*Thread Reply:* Please modify both lines
*Thread Reply:* Apologies for this stupid bug, everyone in the team is currently busy but we will fix it very soon
*Thread Reply:* @Joanna H. Bonczarowska Another known bug that we haven't fixed yet is that one python script is not working with some of the newest python versions. So if you want to run the pipeline soon it would be good to check which python is used in your aMeta conda environment and in case it is a version of python.
If it is a version higher than 3.10, I would remove the conda aMeta environment using the conda command conda env remove -n aMeta
Then open the file aMeta/workflow/envs/environment.yaml and change the line about python with that line: - python>=3.7,<3.10 and recreate the aMeta environment.
*Thread Reply:* Sorry about that, we've been busy indeed and haven't fixed it yet
*Thread Reply:* It now failed at the loading the ncbi.tree, so I will look into the python version
*Thread Reply:* @Joanna H. Bonczarowska does your cluster have internet connection?
*Thread Reply:* Yes it does
*Thread Reply:* I got a new error now 🙈
*Thread Reply:* Please post it here
*Thread Reply:* RuleException: CalledProcessError in file /gpfs/gibbs.../aMeta/workflow/rules/malt.smk, line 26: Command 'source /home/jhb94/.conda/envs/aMeta/bin/activate '/gpfs/gibbs/.../aMeta/.test/.snakemake/conda/45303af74a9961a9c754f37c4bed599c'; set -euo pipefail; python /gpfs/gibbs/.../aMeta/.test/.snakemake/scripts/tmp5bxrmrnn.malt-build.py' returned non-zero exit status 1. File "/gpfs/gibbs/.../aMeta/workflow/rules/malt.smk", line 26, in _ruleBuildMalt_DB File "/home/jhb94/.conda/envs/aMeta/lib/python3.9/concurrent/futures/thread.py", line 58, in run
*Thread Reply:* @Joanna H. Bonczarowska this looks like the wrong python version error. Did you reinstall aMeta from scratch as Zoe suggested?
*Thread Reply:* Could you check (being within aMeta environment) what python --version gives to you?
*Thread Reply:* I did reinstall aMeta and the version 3.9.19
*Thread Reply:* Ok, could you please post the log-file from .test/logs/BUILDMALTDB?
*Thread Reply:* "Output files of filed job BuildMaltDB were removed since they might be corrupted"
*Thread Reply:* no, log-files are not removed, please check the .test/logs/BUILDMALTDB folder. There should be a BUILDMALTDB.log files inside, I would like to look at this file
*Thread Reply:* Version MALT (version 0.6.2, built 12 Sep 2023) Author(s) Daniel H. Huson Copyright (C) 2023 Daniel H. Huson. This program comes with ABSOLUTELY NO WARRANTY. Java version: 20.0.2; max memory: 2.9G Classifications to use: Taxonomy Reference sequence type set to: DNA Seed shape(s): 111110111011110110111111 Number input files: 1 Loading FastA files: 10% 100% (0.3s) Number of sequences: 1,000 Number of letters: 10,000,000 BUILDING table (0)... Seeds found: 9,977,000 tableSize= 16,777,216 hashMask.length=24 maxHitsPerHash set to: 1000 Initializing arrays... 100% (0.0s) Analysing seeds... 10% 20% 30% 40% 100% (2.0s) Number of low-complexity seeds skipped: 43 Allocating hash table... 10% 100% (0.5s) Total keys used: 7,519,628 Total seeds matched: 9,976,957 Total seeds dropped: 0 Opening file: results/MALTDB/maltDB.dat/table0.db Allocating: 41.8 MB Filling hash table... 10% 20% 30% 40% 50% 100% (2.5s) Randomizing rows... 10% 20% 30% 100% (0.3s) Writing file: results/MALTDB/maltDB.dat/table0.idx 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% (2.4s) Writing file: results/MALTDB/maltDB.dat/table0.db Size: 41.8 MB 10% 100% (0.8s) Writing file: results/MALTDB/maltDB.dat/index0.idx 100% (0.0s) Loading ncbi.map: 2,396,736 Loading ncbi.tre: 2,396,740 Loading file: resources/accession2taxid.map
*Thread Reply:* is that all? No more lines in that files?
*Thread Reply:* yes thats all the content of the log file
*Thread Reply:* are you running it on the login-node or on a booked compute node as an interactive job? I mean some login nodes have very little RAM so that even a test-run can't finish successfully 🙂
*Thread Reply:* on a compute node
*Thread Reply:* I will request more memory and try again
*Thread Reply:* to get a clean experiment, could you please just delete the whole results folder as "rm -rf .test/results" and re-run the testrun?
*Thread Reply:* if you booked just one CPU, it might happen it had very little to no RAM. The error you posted above looks like very little RAM (no more than ~50MB) was available at that compute node
*Thread Reply:* booking a lot of RAM is essential for production runs of aMeta, so better to learn from the very beginning how to book more RAM
*Thread Reply:* I am reruning it from scratch with more memory. If it fails again I will request more CPUs.
*Thread Reply:* Ok the error remained, but I got more information on the malt build error.. "java.lang.Out.Of.MemoryError: Java Heap space"
*Thread Reply:* Ok, the solution for Java heapspace error is described here https://github.com/NBISweden/aMeta/tree/main?tab=readme-ov-file#i-get-java-heap-space-error-on-the-malt-step-what-should-i-do
*Thread Reply:* I have seen it before when using malt and could fix it in the "vmoptions" file
*Thread Reply:* ah yes!
*Thread Reply:* Perhaps the easiest solution is to book a few CPUs
*Thread Reply:* fixing the vmoptions should also help but I believe booking more CPU, which automatically should result in more RAM, is an even easier way
*Thread Reply:* It seems like it is running but I dont want to jinx it
*Thread Reply:* I got pdfs generated! It just shows Y. pestis in both samples. Was that the expected output?
*Thread Reply:* Congratulations! You are done with the testrun. Let us try to run a real sample tomorrow
*Thread Reply:* That is great! I needed a "win" today 🙂 Thank you so much for your help. I will try a real sample next
*Thread Reply:* @Nikolay Oskolkov I am now trying to download the datasets to run my samples with the pipeline and I have a question concerning the helping files for building malt repository. When I follow the link ```# Helping files for building Malt database
can be downloaded from https://doi.org/10.17044/scilifelab.21070063
maltntfasta: resources/library.fna maltseqid2taxiddb: resources/seqid2taxid.map.orig maltaccession2taxid: resources/nuclgb.accession2taxid``` It takes me to bowtie2 index for full ncbi nt dataset. Is that correct?
*Thread Reply:* Yes @Joanna H. Bonczarowska, that is correct. Please download only the files that are mentioned above, i.e. you do not have to download the other files from the bowtie2 index
*Thread Reply:* Ah, I see! Thank you
*Thread Reply:* Hi @Joanna H. Bonczarowska, we have recently fixed a few issues with aMeta, it should be more stable now. So if git pull the recent changes, I hope you should not encounter any problems with testrun or real data run. As I said previously, I am ready to assist you to properly install it once so that you can use aMeta in the future for your projects

*Thread Reply:* @Nikolay Oskolkov I finally managed to get the databases downloaded onto the cluster! I also have my first data ready and I will now try to run the pipeline on my real data. For the installation of the job-specific environments part, do I just copy the commands provided on github?
*Thread Reply:* @Joanna Bonczarowska yes, you will need to prepare the config.yaml and samples.tsv files following the instructions from here https://github.com/NBISweden/aMeta?tab=readme-ov-file#quick-start. Please carefully specify the paths to the databases on your disk, and do not forget to unzip the library.fna.gz file (it is a common mistake to keep it zipped). To be on a safe size, you can send me both config.yaml and samples.tsv via a direct message (in order to not overload this thread) so that I could have a look and correct them if necessary. Then you should be ready to start with installation of job specific environments like this: ```cd aMeta
install job-specific environments
snakemake --snakefile workflow/Snakefile --use-conda --conda-create-envs-only -j 20```
*Thread Reply:* Great, thank you very much! I will be in touch with you via private message 😄

Hello! I’m doing sedaDNA metagenomic sequencing from archaeological sites. My focus is eukaryotes (for now, until I can get PhD student to dive further into the data). How much data do you recommend? I’m hoping to go for 10GB for a preliminary look…is that too low though?
*Thread Reply:* Hey @Sarah Martin, Could you give a bit more details on what you want to do ? Non targeted shotgun sequencing of sedaDNA samples won't yield a lot of aDNA, but I'm not sure what you mean by then 10GB. 10 billion reads ?
*Thread Reply:* @Maxime Borry We are taking soil samples down a stratigraphic wall and want to compare the contents and look at the introduction of domesticates, that sort of thing. I’ve done this before with 20Gbases of data, but want to know if there’s a “standard” number of bases, or reads, people use for this sort of thing. I’d ideally like to reduce my sequencing to 10 billion bases (I’m counting in bases because that’s what I order from Novogene, rather than reads). Does that help?
*Thread Reply:* Sounds like a question for @Kevin Nota @Niall Cooke @Merlin Szymanski 🙂
*Thread Reply:* I don't think there is really a standard. We usually aim for 5-10M reads per sample to get an idea of the taxonomic composition. This number of reads tends to give us mainly hits to nuclear genomes and only some mtDNA in the very rich samples. In most cases, I would say doubling or even getting 10 times more data will not help you that much for getting extra taxa. You would get 10 times more reads for the taxa you already have, and some low abundant species that have something like 10 reads will then give you 100 reads, which in most cases is not enough to do things with. In the end, it depends on the sample and budget, if you sequence more, you are likely to get more. I am not sure how to translate this to the number of bases, this would depend on the read length.
*Thread Reply:* @Kevin Nota thanks! This is very helpful. I have another question for you. For my purposes (simply assigning eukaryote taxa) would you suggest Holi? We’ve tried aMeta, but I get the feeling it’s not particularly suited for eukaryotes…
*Thread Reply:* As a start, I think the Holi pipeline is a good choice. We are working on our own pipeline for nuclear DNA that is somewhat inspired by the Holi pipeline. It is hard to recommend a pipeline, that will work well "straight out of the box" and the taxonomic list that these pipelines produce needs validation per site and taxonomic groups. We have been kind of stuck on this part, that comes after mapping and classification - the validation, like how much you can really trust mappings, are the reference genomes that attract mapped reads are good enough? and to what extent is taxonomic bleedthrough a problem etc. 'simply assigning eukaryote taxa' for nuclear data is hard:)
*Thread Reply:* @Kevin Nota thanks! Your input is most appreciated!
*Thread Reply:* @Sarah Martin I agree, while aMeta can technically be used for detecting eukaryotes if you use the full NCBI NT database (prebuilt and provided together with aMeta), it may not be optimal on the authentication analysis step as aMeta's coverage metrics do not currently assume multiple-chromosome reference genomes. I do use aMeta from time to time for environmental / sedimentary aDNA projects for figuring out what organisms are in my samples, however authentication requires some extra work
*Thread Reply:* @Nikolay Oskolkov thanks for your input! We decided to rerun our analyses with HOLI and so far it seems more suitable for our needs. We’ll authenticate the major species separately.
Have folks experience with sending (pathogen) genomic dna for rna bait production e.g. Arbor Bioscience / daicel?
I heard this was cheap way for bait production but there's something about ordering dna of a zoonotic organism that doesn't sound right 😰 or if it was possible to order (from a dna bank..?) and simply deliver straight to bait producer
*Thread Reply:* @Kevin Daly sorry for the late reply just seeing this. I got genomic DNA from several M. tuberculosis strains from https://www.beiresources.org/ I sent the gDNA to Arbor and they created the RNA baits. It’s much cheaper than synthesising baits. The DNA I sent was enough for over 100rxns I believe. The captures worked well. Pretty even and high coverage for good performing samples.
Hi all! I am trying to run gtdbtk classifywf in an assembled dataset (30 MAGs). However, when the analysis arrives to the step "Identifying TIGRFAM protein families" the job fails but I do not have any error in my log file. My guess is that the analysis needs more memory than requested. I am giving 100 Gb and 64 CPUs. Does anyone have experience running this kind of analysis? Do you think the problem could be the memory? This is my command: gtdbtk classifywf --cpus 64 --extension fa --genomedir /data/input --outdir /data/output --skipaniscreen (I also tried to run the analysis without the flag --skipaniscreen but it gave me an error: gtdbtk classifywf: error: one of the arguments --skipaniscreen --mashdb is required)
*Thread Reply:* Ohhh, I just dealt with this and it was a weird conda dependency issue
*Thread Reply:* What version of gtdbtk are you using?
*Thread Reply:* I am using in docker version 2.4.0. Did you solve the problem with the conda dependency?
*Thread Reply:* So it was stupid, it turned out that I also had python (which had gtdbtk) loaded independently of the conda environment I had loaded, so the versions were clashing
another not-stupid-question: for those who have designed/had designed for them in-solution probes for specific genomes, how much tradeoff was given to specificity i.e. excluding probes which may also match environmental relatives?
*Thread Reply:* honestly depends on the species and how many closely related species you are targeting.
*Thread Reply:* but its not just about excluding but also playing with % of allowed mismatches/hybridization temperature/hybridization time for example
*Thread Reply:* Good points. I had been hoping to go along with arbor's recommendation and probe design heuristics but they probably don't take any of these variables into account
*Thread Reply:* They do to some degree if you ask them. Plus they have some set thresholds but some of it doesn't depend on them either since it is dependent on your hybridisation setup during enrichment. Sequence divergence within the species will also be a deciding factor.
ls